This spring semester I am co-teaching a first-year undergraduate course in computer science at Rice University. The name of the course is COMP 182: Algorithmic Thinking. In reality the name of the course can be somewhat misleading, as I would rather title it “Introduction to Discrete Mathematics and Algorithm Design and Analysis”, but it doesn’t quite roll off the tongue as easily. However, this is not a post about the course or teaching experience, this is a post about one of the upcoming topics in the class: counting.
Introduction
What is counting? In general in discrete mathematics counting concerns deriving mathematical expressions which capture the total number of certain objects that exist. Of course, since we can always make an identical copy of an object and thus increase the count by one, what we really mean is counting the objects up to an appropriate notion of isomorphism, or put plainly, counting distinct objects up to whatever notion of being distinct we choose.
While deriving expressions that capture the counts of objects is an important task, what I want to talk about today goes one step beyond the basics of counting, and is rather aimed to provide you with some applied tools you can sue in the future (of course, in the spirit of Alexander Razborov, here by applied I mean applied to the study of mathematics). However, in order to make this primer relatively complete we will start with some basic results in counting, and then switch to the applications of counting techniques and ideas to more interesting problems.
Permutations
This is the classic problem with which most counting modules of discrete math books start: Find the number of ways in which n distinct objects can be arranged in a line. Key information here is that we are talking about distinct objects and an arrangement in a line. It is easy to show that the total number of such permutations is . We can do so by observing that given an ordering of n-1 objects we have n places to put the new object in, and this holds for every ordering of n-1 objects. More formally, if we denote the number of permutations of n objects by then we have . Also, while we are here, it is useful to note that there is exactly one way of putting no objects down at all, or in other words .
Now, we can take a look at the number of permutations in a slightly different way. For the sake of brevity we will be denoting the set of n positive integers as . Consider the set of functions , we will call this set . Recall that a function is an assignment of outputs for any given input, such that exactly one output value is assigned to any input value. Thus, we can see that , as for any given input we have n choices of the output value, and since all choices are independent of each other we have a total of functions. A reasonable follow up question, is to ask how many of those functions are bijections. Recall that we call a function a bijection iff for any two distinct input values it produces distinct outputs, and for every possible output value there exists an input value that maps to it via our function. In other words, when we look at bijections are precisely the functions that assign to each input a distinct output s.t. for any pair of we have . Note, that since the functions are from to it follows that such an assignment has to use all possible output values. If we look closer at the structure of such bijection, we can realize that it is essentially a permutation of n distinct objects, since we can think of input values as positions on the line, and outputs as the distinct objects we place in those positions. Hence, it follows that there is bijections in .
What we saw above is the fundamental use of counting in combinatorics, we find a combinatorial object of interest (say bijections on the set of n elements) and we count the number of such objects. Often we will see seemingly unrelated families of objects that result in the same total counts, offering us different combinatorial interpretations of the same formula. Furthermore, once we are able to count the number of objects with a given property (say being a bijection) among the total collection of objects (say functions between two sets of n objects) we can immediately get the probability of an object selected uniformly at random to possess the property of interest. In the case of the bijections above we have that . We will see why such simple observations can be of use a bit later in the post.
Count once, count twice
Whenever we count number of certain objects we can sometimes derive the formula based on different inputs. This fact becomes useful in cases when we want to show equality between two expressions. In other words, if we want to prove that two expression are equal, one way to do so can be expressing the count of a particular combinatorial object in two ways, each corresponding to one of the expressions. A quick simple example of this proof technique is given by the handshake lemma in graph theory. Consider a simple graph . We want to evaluate the sum of degrees of all vertices . In order to do so, we can think of a counting argument. What is a degree of any given vertex? It is exactly the number of edges incident to that vertex. Hence, if we sum degrees of all the vertices in a graph it follows that we are indirectly counting the edges of the graph. However, each edge in a simple graph is incident to exactly two vertices. This means that when we summed our degrees, we actually counted occurrence of each edge exactly twice (once per endpoint). Thus, it follows that . While this result at first might not appear as a counting argument, if you think about it for a bit you can recognize that we essentially counted the number of edges in a graph in two ways, once by considering degrees of vertices and once, by simply counting all edges.
Another common example for double counting comes from the problem of finding the number of subsets of a set with n elements. Specifically, on one hand each subset is determined by whether each element is in or out of it, i.e. for each element in we have two possible choices either it is in the subset (1) or it is not (0), thus the total number of possible subsets is the number of all possible strings of n characters over the alphabet of , which is . On the other hand, each subset has some size k where , and we know that the number of ways to pick k elements from n is . Hence, we can conclude that .
A fun extension of the argument above can give us a way to count a slightly more complicated set of objects. We are now interested in how many subsets of a set of n elements have an even number of elements in them. On one hand we can express this number as the sum of with , while on the other hand we can consider the following argument: take an arbitrary subset of a set with n-1 elements, now there is a unique way to extend it to an even sized subset of an n element set. Namely, if our starting subset has an even number of elements, we have to exclude, i.e. assign value 0, to the last element in the n element set, if the starting subset has an odd number of elements then we need to add the last element to it to make the size even, i.e. we assign the value of 1 to the last element. It follows that for any of the subsets of a n-1 element set, there is a unique extension to an even sized subset of n element set. Hence, we can conclude that , or in other words, exactly half of all subsets have an even number of elements in them. This is a nice confirmation to an intuitive guess we might have had originally, but sadly the argument does not extend for the number of subsets with a multiple of 3 elements in them. Let’s formulate this more general version of the question. Let be the number of subsets of a n element set that have size divisible by k. We have from our previous work the values , and . It would be nice if was simply , but sadly that is not an integer. However, with a little bit of elbow grease and usage of binomial theorem we can show that . I will leave the solution to this problem as an exercise for a curious reader.
To summarize, we just saw how counting the same combinatorial objects from two perspectives can serve us as a proof technique for showing equalities between expressions. Thus, we are starting to explore the field of applications that counting techniques provide us.
It exists! But I can’t show you an example
Recall how in the bijection counting question we brought up the probability of a randomly picked function from to to be a bijection. Besides the classical uses of probabilities, such as analyses of randomized algorithms, we can also wield probability as a tool for showing existence of objects with desirable properties. This technique is called probabilistic method and it was pioneered in combinatorics by Paul Erdős. The premise for the application is quite simple at the first glance, in order to show that an object with a desirable property exists, we will show that the probability that a random element belongs to the set of elements with desirable property , where is picked from out universe is greater than 0.
In order to make this more concrete we will work through an application of this method to a graph theoretic question. Consider a complete graph on n vertices . We will call a tournament an orientation of the complete graph, i.e. for every edge in a complete graph we will pick a direction and replace it with a directed edge or . It is easy to check that the total number of tournaments on n vertices is . Now, we are interested in determining whether for there exists a tournament with vertices that contains at least Hamiltonian cycles.
To start, consider the sample space of all tournaments on n vertices and assume the uniform distribution on it. Now, we want to introduce a random variable that counts the number of Hamiltonian circuits (recall that a random variable is a function from the sample space to some set, in our case we have ) in a tournament. Fix a node and consider some permutation of the remaining nodes in the tournament (we have a total of such permutations), we want to know when there is a Hamiltonian cycle that achieves that permutation. Clearly we need to have a directed edge from every predecessor to its successor in the cycle, hence in total we need n edges to point in the correct direction determined by our permutation. Now, let’s define an indicator random variable which is equal to 1 whenever the Hamiltonian cycle defined by the permutation exists in a tournament. We want to compute . In order to do so, we note that we need exactly n edges to have fixed orientation, and hence it follows that the probability of such an event occurring is (since we can think of flipping a coin for orientation of each edge, it’s up to reader to check that this process indeed results in uniform distribution over tournaments). Now, by construction we have where goes over all possible permutations on vertices. Hence, by the linearity of expectation we have that . Now, using our previous result we can conclude that . Hence, since the expected value is it follows that there exists some tournament that has at least Hamiltonian cycles in it.
While it is a stretch to claim that this result is purely enabled by our ability to count combinatorial objects, a lot of key ingredients to the proof rely on our ability to count. Furthermore, this showcases the power of probabilistic method as a non-constructive proof technique for showing existence. In other words, if we can properly count the number of certain objects, we may have a hope of proving interesting results about existence of objects with certain properties.
Instead of conclusion
Of course this is a cursory and incomplete account of counting techniques and applications in discrete mathematics. We haven’t touched upon some key counting ideas such as the inclusion-exclusion principle nor did we discuss any of the more advanced approaches such as generating functions. Furthermore, my account of probabilistic method was purposely short and lacked some of the proper rigor required. However, my aim here was to showcase some of the fun applications of counting techniques and to kindle the spark of interest in the reader. I am not certain whether I will write a follow up to this post any time soon, but stay tuned in case if I do.
Cheers, and don’t forget to count your chickens both before and after they hatch! 🐣
This was my first time I did a real hike alone. A bit more on that topic later, but I can say for sure that picking a reasonably well traveled and not too long of a route was a big plus. I got to the trailhead via an Uber and after stepping over a low fence I began the hike at approximately 10:15 AM PST.
Now, in general the Frenchman mountain hike trail can be split into four key areas: the first hill, the “false” peak, the saddle, and finally the summit. Getting to the top of the first hill didn’t take me that long, but in terms of the combined cardio stress (breathing and heartbeat) this was the most gruesome part of the hike. I suspect that this was more of a result of not being warm enough when starting, combined with a rather fast pace I picked up. Funny enough a group of about 6 teenagers was getting off of the hill in the meantime, and two of them managed to go down, back up, and back down in the time I made it to the top. Once at the top of the hill I had another fun encounter this time spotting an older man with two chihuahuas by his side on their way down form the hike. As I’ve learned later they made it all the way to the summit.
The hill is a nice spot to watch planes from a nearby US Air Force base do their training maneuvers. I was lucky during my hike and managed to see a good amount of action, and even snap a few shots of the planes.
After the hill the real hike began. Shortly after starting my trek up to the “false” peak, I met a local hiker A, who briefly explained to me the overall layout of the trail. He mentioned that a lot of people give up after reaching the first peak, since all they see ahead is a descent followed by an even longer ascent to the summit. From there onwards we essentially hiked in parallel with me being first to the “false” peak, and him being first to the real summit. I am almost certain that hiking with A in sight made this a way more fun and easier experience that if I was doing it completely alone. Additionally, I think it helped me maintain a relatively steady pace, not going too fast or too slow through any of the points.
From the “false” peak you get a nice panoramic view towards the north, and as one previous hiker put it “soul crushing” view of the way up about to come towards the south.
As expected the way down from the “false” peak was not much easier than the way up. In general, this is the pattern that held for most of the hike. The way up is strenuous, but the way down is where you are much more likely to fall. Which is not that surprising given that it is way easier to fall down, than to fall up.
Next up on the list was the saddle, a relatively flat area between the two peaks. Since saddle is still elevated compared to the ground level of Las Vegas, there is a nice little window from which you can catch a glimpse of the Strip, as well as mount Charleston’s peaks.
Finally both A and I made it to the summit of the Frenchman mountain. The views from the top were quite amazing, albeit it wasn’t the clearest day, so the city and mountains behind it looked a tad blurry. According to A, who lived in Las Vegas for more than 10 years, and hiked up and down Frenchman mountain at least 7 or 8 times, one of the best views from the summit is at night, when the Strip is lit up with all the neon lights. I wish I was able to see that, but my stay in Las Vegas is short, and I do not have appropriate flashlight gear to trek up a mountain at night.
While we were taking a bit of a break at the top, A explained that if you walk along the fence of the AT&T tower at the top and then do some basic scrambling, you get to see the eastward view from the mountain including lake Mead. We decided to get to that side as well, so we proceeded forward. Surprisingly the narrow passage around the fence is not particularly scary, but the scramble, especially as you get to the top of it, can make your knees feel a bit weak, especially if like me you have a moderate fear of heights. However, the views from that point are absolutely worth a slight tremble in the knees.
At that point A and I parted ways, as I began my journey back, and A decided to chill at the top for some amount of time. The road back was reasonably hard, as the loose rock and gravel did not make for the sturdiest surfaces to walk down a slope on, and the sun was picking up. I was lucky that this march turned out to be colder than usual, but according to A being on the mountain after 11 AM towards the end of the March would not be the smartest move. On my way back, I had solid noon to 1 PM sun blasting, so I had to make sure to reapply the sunscreen or else I’d end up being a fine shade of lobster red by the time I’d be back.
While the planes, the views, and the weather were on my side this time, the local fauna was timid. I only managed to spot a few tiny birds (barely above the size of a hummingbird), but decided not to snap any pictures of them, since they essentially blended with the rocks. The vegetation on the mountain was desert-like as expected.
There were also some cool rocks on the way which seemed to burst from within the other rock formations. They were deep amber in color and a few small fragments I picked up felt almost glass like: relatively light, fragile feeling, yet sharp around thinner edges.
However, since my knowledge of geology and mineralogy is nil, I had no way to identify what those veins were. I probably can Google it, but will rather wait until my parents see the pictures and do the mineralogy Google research for me.
At the end of the hike I felt tired, but accomplished. The solo mission was a complete success, and I enjoyed the views and the challenge. On the way back there were a few moments when as far as I could see I was the only person on the trail. Despite part of my mind thinking about how it is not the best idea to chill in the middle of a desert at noon, I still found it quite meditative to stop in more windy places and take in the vast scale of the nature surrounding me.
I would like to claim that due to my amazing balancing skills and a good choice of hiking boots I managed to stay on my feet for the whole hike. However, such a claim would be false. Towards the end of the hike, as I was descending from the “false” peak, I managed to slip and slide on a bunch of gravel and made full contact with ground. It was a minor and rather graceful fall, so I am not expecting more than a minor bruise on my hip and moderate sized bruise on my ego.
Now, after I made it back to the AirBnB, and am enjoying a nice cold beer, I start feeling my legs and feet drafting a complaint about today’s adventure. Hopefully, I will be mostly recovered by tomorrow as ahead lies an approximately 3 hour drive towards the Zion National Park.
In conclusion, I would recommend the Frenchman mountain trail to anyone who enjoys desert mountain hiking and looks for a moderate hike. Main difficulties of this hike are the steep inclines and loose gravel. However, the overall short length of the trail compensates for it making a round trip in 3.5-4 hours more that achievable, even if you stop frequently for pictures.
On October 16th around 6 AM the plane taking me to Baltimore, Maryland took off from the Hobby airport in Houston. I generally do not mind very early morning flights, and since I still retain the magical (and much envied) ability to sleep on the planes the commute was fine. I arrive in BWI around 10 AM and after picking up my luggage headed directly for the Sheraton Inner Harbor hotel. The hotel did not have an early check-in option available, so I dropped off my bags and went for a walk. I found a neat coffee shop and spent a bit of time catching up on some reading.
Inner harbor in Baltimore, Maryland, around 10:45 AM. Not the best shot, but I managed to catch a bird in this one, so I am posting it.Inner harbor in Baltimore, Maryland, still around 10:45 AM. Still not the best shot, but also I didn’t take any particularly good ones this time. I guess being awake since 3 AM ain’t helpful for my artistic eye.Washington monument at Mt. Vernon in Baltimore, Maryland, circa noon. Quality of the shots improves as the day goes on.
After the coffee break, I met up with Evan Mata and we caught up on school life, travel stories, and I finally got my oyster fix. Two key takeaways from our lunch were as follows: (1) we both wish we took more math courses in college (and in fact this appears to be a common trend for many of my college friends), and (2) Maryland oysters are still delicious, albeit not as cheap as I remembered.
The Local Oyster (Mt. Vernon location). Bloody Mary with an oyster and a dozen of fresh oysters. Hence 13 oysters in total for me, quite a lunch in my opinion.
After the lunch I made it back to the hotel where I finally could check in and catch my breath before the opening keynotes for the ASM NGS 2022. Besides the keynotes, and a brief chat over dinner the first day was not jam packed with activities, so I was able to crash early and catch up on some of the sleep that I missed due to the early flight.
Next day I attended a series of the talks in the morning, after which I headed for the University of Maryland, College Park. UMD campus is quite expansive, and in some strange way it reminded me of the University of Tennessee in Knoxville. The computer science building at UMD is quite impressive both in size and design, and many offices tend to have nice floor to ceiling windows, which is an awesome touch. I did not have too much time to explore the campus, as the visit was relatively tightly packed with meetings, and as soon as those were wrapped up I headed back to Baltimore.
E. A. Fernandez IDEA Factory on the University of Maryland College Park campus. The only shot I took at the UMD campus (sadly, since the campus is quite pretty IMHO).
Although, I unfortunately missed most of the afternoon talks on the first day, I still managed to catch the poster session. In general, I am a big fan of poster sessions. The main benefit of a poster (as opposed to a talk) is the ability to ask a multitude of questions about the nitty-gritty details of the work without feeling guilty of encroaching on other people’s time. I found a good number of quite interesting posters relating to SARS-CoV-2, bacteria and horizontal gene transfer, and even a poster on a speed up for neighbor-joining algorithm. After the poster session I briefly caught up with Mike Nute over dinner which importantly included a crab cake (another checklist food item for me on this trip).
Grabbing dinner with Mike Nute at Phillips Seafood in Baltimore, Maryland. Quite solid crab cake and overall good spread of seafood. Very enjoyable dinner option not too far from the conference hotel location.
The crab cake ended up being quite good, although I have a feeling that with a bit of effort I can probably make a comparable if not a better one at home. However, crab meat is not cheap to come by, so I am not likely to test that hypothesis any time in the near future.
Tuesday was a full conference day for me. I attended all of the talks that day, and tried to keep a list of close notes for most of them. Unfortunately the tables at the conference did not have any outlets or power strips connecting to them or running along the floor. Thus, my note taking was cut short towards the second half of the afternoon, as my laptop ran out of its battery.
The last talk session of they day was a wastewater focused mini-symposium. The talks in this session were quite interesting, and indeed reflected a lot of my experiences and concerns when dealign with wastewater data. Namely, issues of the data quality, metadata annotation, and general complexity of the wastewater samples, which in certain cases ends up being overlooked. I was very happy that I was able to give a talk in this session as well. I was presenting the joint work between Treangen and Stadler labs at Rice University in conjunction with the Houston Health Department. My talk focused on QuaID: a software package we developed for sensitive detection of recently emerged SARS-CoV-2 variants in wastewater sequencing data (preprinted at medRxiv).
My first conference talk looked something like this. ASM NGS 2022, Day 2, wastewater session.Some scallops (which were yummy) at the Watershed in Baltimore, Maryland. Post talk dinner feels good, although sitting on a rooftop terrace might not have been the best decision given it was in the low 50 F range?
Overall the talk went very well, and I enjoyed the experience. Of course, as any public performance giving the talk came with a fair deal of stage fright and nervousness. However, extensive practice runs with my labmates, adviser, and collaborators helped a lot in mitigating anxiety, and polishing the presentation content. As per usual, I do think that several moments could have been executed better on my part, but I’ll hope to use that as the knowledge for preparing my next presentations.
The evening continued on into another poster session with a fair number of exciting wastewater and clinical SARS-CoV-2 work, as well as some benchmarking studies of interest. I felt that the second poster session was a tad bit more lively than the first one, but it also could have been an artifact of my post-talk state. After the poster session, I joined a large-ish group of attendees for dinner and chats. Scallops were not on my Baltimore food checklist, but among the seafood options offered at the Watershed they stood out as an interesting choice. Overall I enjoyed them, although next time I will probably go for the crab cakes.
The last day of the conference was relatively short with only a morning session of the talks. There was a plenty of interesting talks this day, and I was quite pleasantly surprised to hear several talks involving deep learning in a principled and well motivated way. The only drawback of this session was the amount of lightning talks given back to back. It was at times hard to properly note down all the details, and due to the format and the fact that this was the last day it was harder to follow up with the presenters to discuss some of the finer details of their work. However, I still managed to take a good amount of notes, and expand my “papers to read” list by a dozen or so manuscripts.
After the conference, I checked out of the hotel and headed to our last stop before the airport: Johns Hopkins University. First we stopped by the medical campus and grabbed a lunch, which consisted of an absolutely wonderful lamb stew with rice. Then we moved to the Homewood campus pictures of which are attached below.
Afghani food at Kabobi in Baltimore, Maryland near JHU Medical Campus. Amazing lamb stew with veggies, a very hearty and comfortable meal.Johns Hopkins University, Homewood campus, Baltimore, Maryland. Autumn gets to be particularly pretty around this time with all the beautiful colors on the trees.
Being on JHU Homewood campus reminded me of one of the greatest pleasures in the fall season: observing the colorful trees in a light cool breeze. However, similarly to the UMD visit, I did not have much time to wander around and observe the trees, as we mainly focused on the scheduled meetings.
At the end of the trip I was absolutely exhausted, but overall happy with the experience. I managed to tick off several of the key food checklist items on this visit. Of course more importantly, I really enjoyed attending the ASM NGS conference, and finally being able to experience an in-person conference. Giving a talk was a huge added benefit, and all the meetings, lunches, and dinners with multiple new and old acquaintances and collaborators (some of whom I finally got to see in person) made this a very memorable trip.
Getting back to Houston late at night I was reminded that this is a spooky season 🎃!
Hi, if you expected a weekly post here I am extremely glad that you are someone who checks out my blog regularly. However, this week I am skipping my regular post. No, I am not traveling nor am I absolutely crushed by 1001 deadlines. In fact, I had a solid post mapped out and even partially written, but I did not have the energy to fully wrap it up in time.
What’s going to happen next?
First, I am going to be traveling mid of October onwards (within the USA), so catch me on tour (not really). Hence, I am likely to have more travel and conference content rather than the “vignettes” style posts. Nevertheless stay tuned as I will try to provide some form of content be it travel updates or micro-posts at least every Monday.
Second, I am seriously debating taking a tropical vacation. I wish it meant going to an actual tropical resort, but rather I mean a cursory exploration of: (a) tropical geometry (via Maclagan and Sturmfels, 2009); (b) algebraic statistics (via Sullivant, 2018) ; and (c) how all of this relates to bioinformatics (via papers + some of the Lior Pachter’s blog posts). I am not perfectly sure this is the journey I am interested in for the next few months, but it is one of the possibilities I am exploring.
Third, I believe that my most recent posts were avoidant of code snippets which is in part justified by the lack of time to write out meaningful code primers. However, this is not the format I am fully enjoying. I believe that code snippets bring the life to abstract concepts and provide an extra layer of interactive experience that can be beneficial in learning process. Hence, I will be making more effort to bring some code to the posts going forward.
As I mentioned in the previous post, I am taking some time off to figure out which direction to pursue next in the blog post series. However, that does not mean that the whole series itself is going into hiatus. In the meantime, I am going to cover a few other topics in phylogenetics. Additionally, I will cross post some of the one-off posts on Dr. Treangen’s lab website. Now, with logistics out of the way let’s get into tree rearrangements.
Introduction
Whenever we are faced with a problem in combinatorial optimization it is often convenient (or even necessary as the majority of interesting problems turn out to be NP-hard) to come up with some heuristic methods. In particular, we often want to explore a “neighborhood” of an object and locate optimal candidates within this neighborhood after which the search can continue from the new local optima. In order to talk about neighborhoods we need to define a metric on the space of objects we are working with (we talked about metric spaces on phylogenetic trees in severalpreviousposts, but in this case we are focusing solely on the tree topology and associated finite metric spaces). In the context of a search problem it is useful to think about the neighborhood of an object as a set of objects that can be obtained from the initial object via some fixed operation. In phylogenetics such operations on trees are typically called tree rearrangements (Felsenstein, 2004). In this post we will take a look at three classical tree rearrangement operations, but keep in mind that there are other ways to mutate trees which lead to different geometries on the tree space.
In the next three sections we will introduce in detail each of the rearrangement operations in the order of increasing neighborhood sizes considered and then briefly discuss the relationships between these three classes, as well as some implications for computational complexity of related problems.
Nearest neighbor interchange
Nearest neighbor interchange (NNI) operation on a tree consists of picking an internal edge (i.e. an edge that is not incident to a leaf) deleting it along with the four edges incident to the vertices of the original edge, after obtaining four components as the result of the deletion there are three distinct ways of reconnecting them back out of which one yields the original tree. It is easy to see that for an unrooted bifurcating tree on n taxa (i.e. a tree with n leaves) there is a total of n-3 internal edges, and hence a total of 2(n-3) NNI-neighbors. We also note that the operation in this classical sense is defined for bifurcating trees (hence the guarantee that the NNI yields for connected components after the edge removal step). The figure below provides an example of NNI and the resulting tree neighbors.
The original tree is shown in the top left corner and the marked edge is the internal edge along which we perform the NNI. The four connected components labeled A, B, C, and D are shown in the top right corner. The resulting two new trees are shown at the bottom. For each of the three trees we also include the split corresponding to the marked edge.
For a small value of n we can reasonably visualize the adjacency structure on the tree space induced by the NNI operation. Namely we can identify non-isomorphic (read: distinct from the point of view of the properties we care about) labeled tree topologies as vertices and let two vertices be connected by an edge iff we can obtain one topology from another via a single NNI.
An example of adjacency graph on the space of 5 taxa unrooted bifurcating trees under the NNI operation. Original figure and caption are taken from Felsenstein, 2004, p. 40.
In general a phylogenetic tree encountered (or I guess inferred) in the wild might not be bifurcating, so it is quite natural to ask how does one generalize the notion of NNI to an arbitrary labeled unrooted tree. At the core of this operation is the process of picking a distinguished edge and then swapping components resulting from an edge deletion operation. Thus, in the case of a multifurcating tree we simply can do the same operation but with a larger set of neighbors being generated. An example is provided in the figure below.
A few possible neighbors of the original multifurcating tree (top left) under the NNI on trees with varying internal node degrees. Note that in the case where we allow multifurcations we ought to be careful in the way we define the NNI operation as some interchanges can be problematic (consider what happens if we reconnect 5 components into a star graph).
There is some amount of work on the NNI operations and NNI induced metric on phylogenetic trees (Hon and Lam, 1999), although the area tends to feel relatively sparse in terms of the research done.
Subtree prune and regraft
Subtree prune and regraft (SPR) operation on a tree consists of snipping off a branch (which can be an internal or external edge) and then inserting the snipped off subtree onto one of the edges of the remaining tree. This operation is quite well illustrated by its name, especially if you ever had to deal with grafts on actual trees (my grandfather loved to experiment with fruit tree grafting, which to his credit managed to give us a wonderful apricot half-tree on a wild apricot that used to grow in the garden). The figure below is an example of a SPR operation performed on a tree.
A subtree is snipped off from the original tree along the edge marked in red (top left) and then re-attached to the remaining tree on an edge marked in orange. It can be helpful to think of this operation as temporarily turning one of the subtrees into a rooted tree and then reinserting its root into the remaining tree.
It is easy to see that SPR operation results in a larger neighborhood set than the NNI operation. It also useful to note that any tree T’ that can be obtained from a tree T via a single NNI can also be obtained via a single SPR. In other words, if we define the set NNI(T) of trees that can be obtained from T via a single NNI, and the set SPR(T) of trees that can be obtained from T via a single SPR, then the following inclusion holds: NNI(T)⊆SPR(T) (Maddison, 1991). In the spirit of ancient geometers instead of a formalized proof, we will provide the following figure and leave it up to the reader to convince themselves that the inclusion above is indeed true.
Similarly to the NNI case we can compute the exact number of neighbors under the SPR operation for an unrooted bifurcating binary tree on n taxa. The size of this neighborhood similarly to the NNI case is independent of the topology of the tree T and is equal to 2(n-3)(2n-7) with a detailed counting argument provided by Allen and Steel, 2001 on p. 4.
We also note that the way in which we graft a subtree back in will always create an internal node of degree 3, so without a modification to this operation the question of generalizing it to multifurcating trees can be ill-posed.
Tree bisection and reconnection
Tree bisection and reconnection (TBR)operation is the most expansive operation among the three we are considering in this post, in the sense that it yields the largest neighborhoods. TBR consists of splitting the original tree T into two subtrees T’ and T” along a branch and then reconnecting them by joining any two branches of T’ and T” via an edge (in case if one of the subtrees is just a leaf the leaf is simply joined back to the tree via a new branch; in all cases vertices might have to be added to maintain a proper bifurcating tree). The figure below illustrates a TBR operation being performed.
The original tree is shown in the upper left corner. The edges to be joined are marked in orange (bottom), and the new joining edge is marked in red (bottom right). Note, that the tree obtained after the specified TBR operation is isomorphic to the tree obtained via SPR with regrafting onto the orange edge of the bottom left subtree.
The inclusion we observed for the NNI and SPR neighborhoods extends further and the following holds: NNI(T)⊆SPR(T)⊆TBR(T) (Maddison, 1991). Furthermore, by noting that any TBR can be thought of as a two step process where we first re-attach T” onto the specified edge of T’ and then reposition the attached T’ in order to get correct edge match, we can show that any TBR operation can be replicated by at most two successive SPR operations.
Finally, we note that unlike NNI and SPR, the TBR neighborhood size does depend on the topology of the tree T. However, and upper bound on the size of the neighborhood can be computed and is given by (2n-3)(n-3)2 (Allen and Steel, 2001, p.5).
Induced metrics and computational (in)tractability
We already noted in the introduction that the tree rearrangement operations induce corresponding metrics on the space of phylogenetic trees (more precisely in our current case: the space of unrooted bifurcating tree topologies). We will denote these metrics dNNI(T, T’), dSPR(T, T’), and dTBR(T, T’), respectively. From the inclusion relation on the neighborhoods it immediately follows that for any two unrooted bifurcating trees we have the following inequality: dTBR(T, T’)≤dSPR(T, T’)≤dNNI(T, T’). Furthermore, from our observation in the previous section we also can conclude that dSPR(T, T’)≤2×dTBR(T, T’).
Although we have provided an explicit example of the adjacency graph under NNI metric for the space of unrooted bifurcating trees on 5 taxa, it is not immediately obvious that in the general case the NNI adjacency graph will be connected (i.e. we are not a priori guaranteed to have a connected metric space). However, it turns out that indeed the adjacency graph under the NNI metric is indeed connected, and hence the metric space induced by dNNI(T, T’) is connected. It is easy to see that as a corollary we also get connectedness for the SPR and TBR spaces.
Since the metric spaces are connected and finite we can ask what are their respective diameters (i.e. the smallest distance between two points furthest apart in the space; this is the same definition as the graph diameter). An interesting non-trivial bound on the diameter of the NNI space was derived by Li et al., 1996: authors show that the diameter of the NNI adjacency graph (which we will denote as ΔNNI) is bounded below and above by nlogn terms. More precisely, the following inequality holds: . Allen and Steel, 2001 extend this result to the SPR and TBR cases giving the following inequalities: (a) n/2-o(n)≤ΔSPR≤n-3; (b) n/4-o(n)≤ΔTBR≤n-3.
However, knowing the diameter of a space does not imply that we can easily compute the distances within it (i.e. shortest paths via NNI, SPR or TBR operations from one tree to another). In fact, the problems of computing NNI, SPR or TBR distance between two arbitrary unrooted bifurcating trees are all NP-hard (NNI: DasGupta et al., 2000; SPR: Bordewich and Semple, 2005, Hickey et al., 2008; TBR: Hein et al., 1996). Allen and Steel, 2001 show that the SPR distance is fixed-parameter tractable, but in general the fixed-parameter tractability results in algorithms that can work efficiently only on small trees or pairs of trees which are relatively close to each other (for a more principled discussion see Whidden and Matsen, 2018). In general, it appears to be a rare case for a metric that is biologically interpretable to be efficiently computable, and vice-a-versa (take for example the Robinson-Foulds distance which is efficiently computable, but not biologically well grounded). There are some recent attempts at developing variants of tree rearrangement operations that have both biological interpretability and are computationally tractable (Collienne and Gavryushkin, 2021).
While we could possibly discuss other tree rearrangement operations or explore the discrete geometries arising from the ones we mentioned in more detail, such work would likely require multiple posts. Thus, we will wrap up here and invite the reader to continue the exploration via following the links provided in the references section.
Data and code availability
All illustrations with the exception of the one from Felsenstein, 2004 were made with draw.io. No additional materials or code were used in this post.
What’s next
I am still deliberating on the exact direction to take for the next stretch of posts, so in the meantime I am intending to continue the one-off posts where I pick a random, but interesting to me, topic in mathematical phylogeny and try to write a coherent text about it. As per usual, if you are interested in me writing about a specific topic do not hesitate to reach out.
References
Allen, Benjamin L., and Mike Steel. “Subtree transfer operations and their induced metrics on evolutionary trees.” Annals of combinatorics 5, no. 1 (2001): 1-15. https://doi.org/10.1007/s00026-001-8006-8
Bordewich, Magnus, and Charles Semple. “On the computational complexity of the rooted subtree prune and regraft distance.” Annals of combinatorics 8, no. 4 (2005): 409-423. https://doi.org/10.1007/s00026-004-0229-z
Collienne, Lena, and Alex Gavryushkin. “Computing nearest neighbour interchange distances between ranked phylogenetic trees.” Journal of Mathematical Biology 82, no. 1 (2021): 1-19. https://doi.org/10.1007/s00285-021-01567-5
Gupta, Bhaskar Das, Xin He, Tao Jiang, Ming Li, and John Tromp. “On computing the nearest neighbor interchange distance.” In Discrete Mathematical Problems with Medical Applications: DIMACS Workshop Discrete Mathematical Problems with Medical Applications, December 8-10, 1999, DIMACS Center, vol. 55, p. 125. American Mathematical Soc., 2000. PDF on author’s page
Hein, Jotun, Tao Jiang, Lusheng Wang, and Kaizhong Zhang. “On the complexity of comparing evolutionary trees.” Discrete Applied Mathematics 71, no. 1-3 (1996): 153-169. https://doi.org/10.1016/S0166-218X(96)00062-5
Hickey, Glenn, Frank Dehne, Andrew Rau-Chaplin, and Christian Blouin. “SPR distance computation for unrooted trees.” Evolutionary Bioinformatics 4 (2008): EBO-S419. https://doi.org/10.4137/EBO.S419
Hon, Wing-Kai, and Tak-Wah Lam. “Approximating the nearest neighbor interchange distance for evolutionary trees with non-uniform degrees.” In International Computing and Combinatorics Conference, pp. 61-70. Springer, Berlin, Heidelberg, 1999. https://doi.org/10.1007/3-540-48686-0_6
Li, Ming, John Tromp, and Louxin Zhang. “On the nearest neighbour interchange distance between evolutionary trees.” Journal of Theoretical Biology 182, no. 4 (1996): 463-467. https://doi.org/10.1006/jtbi.1996.0188
Maddison, David R. “The discovery and importance of multiple islands of most-parsimonious trees.” Systematic Biology 40, no. 3 (1991): 315-328. https://doi.org/10.1093/sysbio/40.3.315
Whidden, Chris, and Frederick A. Matsen. “Calculating the unrooted subtree prune-and-regraft distance.” IEEE/ACM transactions on computational biology and bioinformatics 16, no. 3 (2018): 898-911. https://doi.org/10.1109/TCBB.2018.2802911
I am currently deciding on the direction this post series will take after next week. Please check out the What’s next section and let me know if you have any preferences among described routes.
Recap
In the previous post (following Gavryushkin and Drummond, 2016) we have defined a geometry on the space of phylogenetic ultrametric trees and called the resulting metric space 𝛕-space. We also briefly mentioned the geometry on the space of trees defined by Billera, Holmes and Vogtmann, 2001, and referred to the resulting construction as BHV space.
In this post we will provide a brief overview of the algorithm presented in Owen and Provan, 2010, which allows us to compute the geodesics in 𝛕-space and BHV space in polynomial time (in the number of the tree leaves/taxa). For the ease of exposition we will delegate detailed overview of the BHV space to a later post. Hence, in what follows we will assume without the proof that BHV space is a CAT(0) complex and trees in BHV space are parametrized by the internal branch lengths with different orthants corresponding to different tree topologies.
Goal
As we mentioned before one of the key properties we want from a metric space structure on the tree space is the efficient computation of geodesics. Clearly given any finite metric space (complexes constructed in BHV or 𝛕-spaces are not finite, but the topologies are and the problem of distance within a fixed orthant determined by topology is easy to solve) we can simply consider all possible paths and then pick the one minimizing the length function. However, such approach yields an algorithm that will fail to finish computations even for modestly sized spaces.
Thus, our goal is to have an efficient algorithm that can find shortest path in polynomial time with respect to the number of leaves our trees can have.
CAT(0) properties
Since both BHV and 𝛕-spaces are CAT(0) we can leverage nice properties of the non-positively curved geometries to get an idea for an efficient algorithm. The key idea behind the algorithm comes from the following lemma (Lemma 2.1 in Owen and Provan, 2010, and Proposition 1.4, p. 160 in ).
Lemma 1.In CAT(0) space, every local geodesic is a geodesic.
Proof. Consider a local geodesic between points and . Choose a set of points such that . Note that it follows from the definition of a local geodesic that is a geodesic. Now, assume by induction that is a geodesic for all $j\leq i$. Hence, we have by the induction hypothesis and as noted above. Consider a corresponding model triangle in Euclidean space (if you forgot what a model triangle is check: Bridson and Haefliger, 2013 p. 158 or previous post on CAT spaces). Let be a point on such that . Since we are working in a CAT(0) space it follows that . On the other hand, by induction hypothesis we have . Combining this result with triangle inequality gives us , and hence and therefore . From here we can conclude that is a geodesic and hence $\Gamma_\varepsilon$ is a geodesic.
Thus, the main idea used in the paper is to start with any path between two trees (consider the path from a tree to the star tree and then back to target tree, this path called cone path is a common idea in the analyses of tree spaces; recall: Robinson-Foulds metric) and iteratively check whether it’s a local geodesic refining the path in cases when that’s not the case.
Path space geodesics
Following Owen and Provan, 2010 we will assume that the trees between which we are computing geodesics have distinct edges. We will also call two sets of edges and compatible if all pairs of splits associated with these edges are compatible, or equivalently if the union defines a tree. In other words, compatible sets of edges produce “intermediate” trees between and . Now, for a pair of partitions , of and , respectively, we can define an associated path space given that the following property holds: for every , and are compatible. To construct the path space consider the collection of orthants given by , where is the mapping of a tree to the corresponding orthant. We will call the corresponding partition pair the support, and the shortest path between and in the path space geodesic. These definitions make the geodesic in the tree space a more concrete and constructable object, which of course is important for any algorithmic approach. Furthermore, the following theorem from Billera, Holmes and Vogtmann, 2001 (see Proposition 4.1) guarantees that the path space geodesics are recovering the true geodesics.
Theorem 1. For disjoint trees the geodesic is a path space geodesic for some choice of .
We will also need the following two results, the first of which is proven in Owen, 2011 and the second in Owen and Provan, 2010 (Theorem 2.5).
Theorem 2.Let be a geodesic between and . Then there exist partitions latex \mathcal{B}=\{B_1,…,B_k\} of and , respectively, such that (a) form a support of path space in which is a path space geodesic, and (b) the following condition is satisfied:
.
A path space satisfying the above condition for its support is referred to as aproper path space, and the corresponding path space geodesic is called a proper path.
Theorem 3.A proper -path with support is a geodesic iff for each pair of edge sets in the support there is no nontrivial partition , such that is compatible with and .
Theorem 3 gives us the final ingredient needed to build out an algorithm for finding BHV space geodesics. Namely, partitions of edge sets give us a way to construct proper paths and the condition on local improvement of such proper paths guarantees that we achieve a geodesic.
Algorithm overview
As we mentioned earlier the natural choice for the starting path is the cone path between the two trees and , more formally it’s a proper path space defined by the support and the corresponding path is given by uniform contraction of all edges which results in star tree and then a follow up uniform decontraction. Now, the algorithm will proceed iteratively at each step checking whether the condition in Theorem 3 is satisfied and if not proposing a new support and a new proper path.
In order to facilitate an efficient solution for the iterative step, the problem of checking the condition of Theorem 3 and finding a new path can be recasted as an independent set problem over a bipartite graph. Namely, we will construct the incompatibility graphlatex A\subseteq E(T), B\subseteq E(T^\prime)$ as a bipartite graph with vertices given by and edges given by the pairs where the splits associated with and are incompatible. It is easy to see that for any compatibility of and is equivalent to forming an independent set in $G(E(T), E(T^\prime))$.
Thus, the condition from Theorem 3, can now be recasted as follows: Given two edge sets $A\subseteq E(T), B\subseteq E(T^\prime)$ does there exist a pair of partitions , such that the vertex set is independent in and
.
In particular, a proper path with support is geodesic iff no edge set pair satisfies the above condition.
Now, since the complement of an independent set is a vertex cover we can reformulate the above problem as a minimum weight (by transforming the inequality condition) vertex cover problem, which can be solved on bipartite graphs in . Further analysis of the algorithm steps yields that the total runtime to find the geodesic is . For the exact details of the proof of correctness of the algorithm and the runtime I suggest reading the original manuscript by Owen and Provan, 2010.
Data/code availability
This post has no associated custom code or data. Those looking for an implementation of the algorithm described should check out Megan Owen’s webpage or GitHub repo.
What’s next
This post concludes the beginning of our exploration of the geometry of the space of phylogenetic trees. There are several ways we can proceed from here.
First, we can finally start tackling the foundational paper of Billera, Holmes and Vogtmann, 2001. This path will likely require us to spend a few posts dissecting original BHV space and some associated results from metric geometry and CAT(0) spaces. We then can return to the paper of Gavryushkin and Drummond, 2016 and explore the t-space construction in a way similar to our exploration of the 𝛕-space. Finally, we can then continue our journey by catching up to more recent work by Megan Owen et al. on defining and computing statistics in BHV space (Brown and Owen, 2020), comparing phylogenetic trees with different leaf sets (Grindstaff and Owen, 2018), and extending the framework to phylogenetic cactuses (a type of phylogenetic network, Huber et al., 2021).
Second, we can instead pivot completely and ask a general question of what other geometries one can consider on the space of phylogenetic trees. In particular, we can take a look at algebraic geometry inspired view of the phylogenetic tree space as a tropical geometry (Monod et al., 2018), and an information geometry angle which gives rise to wald space (pun credit to the authors) of the phylogenetic trees (Garba et al., 2021). I will most likely skip the category theoretic angle for the topic (Baez and Otter, 2015), since frankly speaking I have no desire to introduce the notion of an operad.
Third, we can temporarily halt the process of investigating deeper into the various geometries on the tree space and ask more broadly what are geometries of interest in the case of phylogenetic networks. This route will require us to go back to some definitions of the phylogenetic network and related concepts, and will likely demand at least some amount of rehashing of the biological context.
Since, I am still debating which route is of the most interest to me at the moment, next week’s post will likely be sparser than usual, as I am figuring out which rabbit hole to pick as the next destination. However, if you actually would prefer me to pick one of these three directions do not hesitate to comment or reach out to me via other means of contact.
References
Baez, John C., and Nina Otter. “Operads and phylogenetic trees.” arXiv preprint arXiv:1512.03337 (2015). arXiv:1512.03337
Billera, Louis J., Susan P. Holmes, and Karen Vogtmann. “Geometry of the space of phylogenetic trees.” Advances in Applied Mathematics 27, no. 4 (2001): 733-767. https://doi.org/10.1006/aama.2001.0759
Bridson, Martin R., and André Haefliger. Metric spaces of non-positive curvature. Vol. 319. Springer Science & Business Media, 2013. https://doi.org/10.1007/978-3-662-12494-9
Garba, Maryam Kashia, Tom MW Nye, Jonas Lueg, and Stephan F. Huckemann. “Information geometry for phylogenetic trees.” Journal of Mathematical Biology 82, no. 3 (2021): 1-39. https://doi.org/10.1007/s00285-021-01553-x, arXiv:2003.13004
Gavryushkin, Alex, and Alexei J. Drummond. “The space of ultrametric phylogenetic trees.” Journal of theoretical biology 403 (2016): 197-208. arXiv:1410.3544
Grindstaff, Gillian, and Megan Owen. “Geometric comparison of phylogenetic trees with different leaf sets.” arXiv preprint arXiv:1807.04235 (2018). arXiv:1807.04235
Huber, Katharina T., Vincent Moulton, Megan Owen, Andreas Spillner, and Katherine St John. “The space of equidistant phylogenetic cactuses.” arXiv preprint arXiv:2111.06115 (2021). arXiv:2111.06115
Monod, Anthea, Bo Lin, Ruriko Yoshida, and Qiwen Kang. “Tropical geometry of phylogenetic tree space: a statistical perspective.” arXiv preprint arXiv:1805.12400 (2018). arXiv:1805.12400
Owen, Megan. “Computing geodesic distances in tree space.” SIAM Journal on Discrete Mathematics 25, no. 4 (2011): 1506-1529. arXiv:0903.0696
Owen, Megan, and J. Scott Provan. “A fast algorithm for computing geodesic distances in tree space.” IEEE/ACM Transactions on Computational Biology and Bioinformatics 8, no. 1 (2010): 2-13. https://doi.org/10.1109/TCBB.2010.3
In the previous post we have introduced the notion of a CAT(0) space, and briefly talked about some of the properties of CAT(0) spaces, and in particular cubical complexes. In this post, we will take this knowledge a step further into the applied direction of phylogeny. Following Gavryushkin and Drummond, 2016 we will define a metric on the space of ultrametric phylogenetic trees, show that the resulting space is CAT(0), and then discuss some of the implications of these results. Similarly to the previous post, we will have quite a bit of mathematical notation to deal with, so I will attempt to use visual aids whenever possible to improve exposition.
Motivation
One way of defining a metric on a space of phylogenetic trees is to embed the tree space into some metric space . Thus, by associating every tree in the tree space with some point in the chosen metric space, we induce a pseudometric on tree space. However, such embeddings/parameterizations are not guaranteed to be “nice” with respect to the questions we aim to answer. Namely, not all embeddings are injective (multiple distinct trees can end up being mapped to the same point in , hence in general we induce a pseudometric rather than a metric via embedding) although in the case of Gavryushkin and Drummond, 2016 the embedding has to be injective by definition. Additionally, not all embeddings are surjective (meaning that a path in might contain non-tree associated points) which creates a plethora of problems ranging from potential multiple distance minimizing solutions to the lack of tree space midpoints.
Thus, it is natural to formulate a list of desiderata for the embedding that would enable fruitful analyses. From now onwards (unless otherwise specified) we will assume that the embedding is injective.
In order to make continuous distributions behave correctly under the pullback into the tree space, we have to require the image of the embedding to be path-connected. Note, that having a path connected image is also intuitively “nice” as the continuous tree transformations would result in a path being traced out in the model metric space. Hence, we have:
(D1)The set is path-connected in .
However, being path-connected does not guarantee that the shortest path between the two points will lie within the image. Hence, we also want:
(D2) The set is convex in .
We note that the injectivity criterion forces a lower bound on the dimension of . However, if we want to define probability measure on that can be meaningfully pulled back onto we need to ensure that:
(D3) has the same dimension as .
Next, for statistical analyses to stay sound we need to ensure uniqueness of the shortest paths. This is the point in our desiderata list where you might have a brief flashback to the CAT(0) space discussion from previous week.
(D4) The space is uniquely geodesic.
Finally, since we are ultimately tackling these questions from the computer science viewpoint, we have the last desiderata that ties this into the computational realm. Namely, we want the following:
(D5) Geodesics in are computable.
In reality we probably want an even stronger version of (D5), which is:
(D5′) Geodesics in are efficiently (i.e. polynomial time) computable.
It’s worth remarking that in a general metric space we do not necessarily have (D5) with a simple example being the halting problem reduction to shortest paths in an infinite graph.
It is also important to realize that these desiderata do not guarantee existence of such embeddings, and are rather criteria for discerning more or less useful parameterizations of the tree space.
A quick aside
In general, we can attempt to parametrize the whole tree space (that’s precisely what Billera, Holmes and Vogtmann, 2001 do in their paper, which lies at the foundation of the connection between the geometry of non-positive curvature spaces and phylogenetic trees; we will refer to this space as the BHV space going forward) or we might focus on a specific subclass of trees such as ultrametric phylogenetic trees (recall that a tree is ultrametric is the distance from root to every leaf is the same). The caveat is that a parametrization that enjoys our desiderata (D1)-(D5′) for the whole tree space can fail to do so when restricted to the space of ultrametric trees. This is precisely the case for the BHV space parametrization. Hence, it can be of interest (motivated by the evolutionary models context) to explore parametrizations designed specifically for the space of the ultrametric trees. This is precisely what is done in Gavryushkin and Drummond, 2016, and what we are aiming to do in this post.
𝛕-space
We now will construct one version of a space of ultrametric trees called the 𝛕-space. To proceed consider an ultrametric tree T on n taxa, with internal nodes ordered by their time from the extant taxa. We will then parametrize the tree by mapping it to its ranked topology and a n-1-dimensional vector , where $\tau_i$ is the time difference between the i-th and i+1-th nodes. The picture below taken from Gavryushkin and Drummond, 2016 represents this parametrization for a tree on 5 taxa.
Hence, formally we have the mapping given by that embeds the space of ultrametric phylogenetic trees into a disjoint union of (Semple and Steel, 2003) non-negative real n-1-dimensional orthants (i.e. sets of the form ). We will impose an upper bound on all orthants to turn our construction into a cubical complex for the ease of the exposition. However, the construction of 𝛕-space with orthants will still yield a CAT(0) space with the key properties and desiderata (D1)-(D5′) preserved.
Note that the ranked topology in this case is a stricter condition on the shape of the tree than just the topology constraint. In particular, the two trees pictured below have distinct ranked topologies, and hence will belong to two distinct orthants in the 𝛕-space.
Figure 2. An example of two distinct ranked topologies.
Finally, to properly turn this into a cubical complex we will need to identify the isometries that glue the faces of our cubes together. However, this is rather obvious by construction, since whenever any 𝛕 coordinate becomes 0 we observe a collapse of two nodes in the ranked hierarchy to the same level. Hence, for example the two trees shown above will belong to the cubes that share the face. Since the resulting space is a cubical complex, we can naturally consider the Euclidean metric within each cube with paths for joining points in different cubes being the sums of the respective within cube paths.
The clip below shows how a tree can vary in 𝛕-space while being restricted to a single orthant determined by its ranked topology. Note that just as we discussed above in order to stay within the orthant in 𝛕-space we must keep the ranked topology constant.
For comparison, we show how a similar ultrametric tree can vary within a single fixed orthant of a BHV-space. In this case, the topology of the tree is fixed, but the ranked topology can vary.
Properties
After defining the 𝛕-space it is natural to ask whether it manages to achieve our desiderata. In order to check these properties we need to first figure out how many common faces two cubes in our complex can share (recall from the previous post that a theorem of M. Gromov gives characterization of CAT(0) cubical complexes in terms of face-sharing properties).
In general, similarly to the previous post by face of a cube we mean any sub-cube, thus it is useful to have a separate term for faces of codimension 1 (i.e. faces that are 1 less dimensional than the cubes). Hence, we will call faces of codimension 1 facets. In particular, any facet can be shared by 1, 2 or 3 cubes in total. If we let , then the resulting facet is only contained in the cube of corresponding rooted topology. If setting does not result in a multifurcating topology then the corresponding facet will be shared by exactly two cubes (see example in Figure 2 and/or Figure 2 of Gavryushkin and Drummond, 2016). Finally, if we get a multifurcation then the total number of cubes sharing the facet is 3.
Now, we are ready to take a stab at the critical result about the 𝛕-space, namely that it is a CAT(0) space, and hence uniquely geodesic. We will recall the theorem of M. Gromov here to reiterate the key result we need to prove.
Theorem 1. A cubical complex with the intrinsic Euclidean metric is CAT(0) if and only if is connected, simply connected, and for all natural : if three -cubes of share a common -cube and pairwise share common distinct -cubes, then they are contained within a -cube in .
We start by noting that the metric we introduced on our cubical complex is precisely the intrinsic Euclidean metric, and it is easy to see from our construction that the resulting complex is connected and simply connected.
To finalize the proof we need to introduce the concept of the link of a vertex v in a complex. We say that the graph G with nodes given by facets containing v and edges given by cubes that contain the two incident nodes as facets is the link of the vertex v.
First, we need to show that the cubes of the dimension (k+2) in the theorem cannot be the highest dimensional cubes in the complex. If that was the case, then the link of origin would have to contain a 3-cycle (given by the pairwise distinct (k+1)-cubes) which contradicts the result that the nearest-neighbor interchange graph does not contain 3-cycles. Thus, it follows that each of the (k+2)-cubes has at least one coordinate equal to 0. For the sake of clarity of the presentation we will assume that this is unique coordinate for each of the cubes, although the similar argument can be made in the general case. Let i, j, and r denote the respective coordinates in three cubes. We have three cases to analyze:
(i=j=r) This is impossible due to no 3-cycle property of the link of the origin.
(i=j) In this case the first two cubes must share a (k+1)-cube, and since all shared (k+1)-cubes must be pairwise distinct it cannot be the cube obtained via setting . Hence, there exists s.t. it is greater than zero in both of the first two cubes, and is zero in their shared (k+1)-cube. Now, if the first and third cube share a (k+1)-cube then it follows that their i and r coordinates both have to be zero in the shared cube, implying that the s coordinate has to be resolved in the same way between the first and third cube. However, the same exact argument can be repeated for the second and third cube implying that the first and second cubes had to be indetical.
(all distinct) In this case the left out coordinate (r for the first and second cube, i for the second and third, j for the first and third) has to be resolved the same way in the two cubes under consideration. Now, we can construct a (k+3)-cube that contains all three cubes by taking the first cube and resolving its zero coordinate (i) the same way as it is resolved in the remaining two cubes.
This analysis concludes the proof of the property required to claim that the cubical complex we defined is indeed CAT(0). It immediately follows that the geodesics in 𝛕-space are unique. At this point we are essentially done with (D1)-(D4) and the only remaining piece is the (efficient) computability of the said geodesics.
Due to the constraints on the length and scope of the post we will not dive into how does the algorithm of Owen and Provan, 2010 work in the 𝛕-space. Instead, we will discuss the algorithm in its own dedicated post.
Data and code availability
Code used to generate the 𝛕-space and BHV-space tree examples, as well as HTML outputs from Bokeh are provided in the following archive.
We spent a noticeable amount of time discussing mathematical properties of different metrics that can be imposed on tree spaces. As our next step, we will switch the lens to a computational perspective and explore the algorithm proposed by Owen and Provan, 2010. We might also touch upon more recent work in the area of the efficient computation of geodesics in CAT(0) cubical complexes. Once we are satisfied with our algorithmic solutions, we will continue the journey by exploring the BHV-space and t-space of phylogenetic trees.
References
Billera, Louis J., Susan P. Holmes, and Karen Vogtmann. “Geometry of the space of phylogenetic trees.” Advances in Applied Mathematics 27, no. 4 (2001): 733-767. https://doi.org/10.1006/aama.2001.0759
Bridson, Martin R., and André Haefliger. Metric spaces of non-positive curvature. Vol. 319. Springer Science & Business Media, 2013. https://doi.org/10.1007/978-3-662-12494-9
Gavryushkin, Alex, and Alexei J. Drummond. “The space of ultrametric phylogenetic trees.” Journal of theoretical biology 403 (2016): 197-208. arXiv:1410.3544
Owen, Megan, and J. Scott Provan. “A fast algorithm for computing geodesic distances in tree space.” IEEE/ACM Transactions on Computational Biology and Bioinformatics 8, no. 1 (2010): 2-13. https://doi.org/10.1109/TCBB.2010.3
Semple, Charles, and Mike Steel. Phylogenetics. Vol. 24. Oxford University Press on Demand, 2003.
Unlike the previous two posts it will not be obvious how what we will discuss connects to phylogeny. Furthermore, we will be diving into a noticeably more mathematically dense content this time. I will try to use some analogies and visual aids through out this post in order to make the material slightly more accessible.
Introduction
Geometry is a wonderful subject (even if I struggled with it in high school) that makes appearances in many aspects of various sciences (including social ones). However, musing about geometry at large is better left to people who are (a) more proficient in the study of it, and (b) have an acumen for writing more lengthy works, hence I will simply point you in the direction of Jordan Ellenberg’sShape. What we will concern ourselves with today is a more specific slice of the geometry in which we will look at metric spaces of non-positive curvature. I will not define the notion of a metric space again, as you can simply navigate either to the previous post in the series or Wikipedia. The bulk of this post will consist of defining non-positive curvature, and then we will follow up with a few properties of such spaces, and some examples.
Geodesics are a natural notion extending the concept of the “shortest” path curve to general metric space framework. More precisely, given a metric space a geodesic from to is the map s.t. and . We will call a metric space a geodesic space if for any there exists a geodesic from to . Furthermore, if for all pairs of points such geodesic is unique we will call such space uniquely geodesic (see Bridson and Haefliger, 2013, pp. 4-8 for additional details and examples).
For example, the classic Euclidean space is uniquely geodesic with the geodesics given by straight lines, i.e. for any two points we have the geodesic given by . On a sphere geodesic between two points is the arc segment obtained by intersecting a plane through , and the center of the sphere with the surface (i.e. the great circle arc). An example of a geodesic triangle on a sphere is given below (image courtesy of Wikipedia).
Note, that a sphere is not a uniquely geodesic space, since any pair of diametrically opposed points has an infinite set of possible geodesics between them.
Triangles
Just like the notion of a geodesic extends our naïve understanding of the shortest paths, the notion of a geodesic triangle (Bridson and Haefliger, 2013, p. 158) extends our notion of a triangle. Given a metric space a geodesic triangle is a set of tree points called vertices and a choice of three geodesic segments joining them called sides, we will denote such a triangle or more briefly . Note, that the later notation is not precise, as in the case of a non-uniquely geodesic space there is not necessarily a unique choice of a geodesic between two vertices. Finally, we will write to indicate that belongs to the union .
In order to define a CAT(k) space we also need the notion of a comparison triangle. In order to avoid a lengthy (and somewhat involved) discussion of the nuances about the model spaces and corresponding notation, we will focus on three general types of model spaces: Euclidean n-dimensional space, the n-sphere, and hyperbolic n-space. We already defined geodesics in the case of . For the n-sphere we will define the metric via cosine distance where the inner product is in the corresponding embedding and the corresponding geodesics are given by minimal great arcs (for full definition and description see Bridson and Haefliger, 2013, pp. 16-17). For the hyperbolic n-space we will use the same construction as Bridson and Haefliger, 2013 (p. 18-20) by considering the space which consists of with bilinear form and defining . The distance on this space will be given by (similarly to the sphere case, see Bridson and Haefliger, 2013, pp. 18-23 for details).
Finally, a comparison triangle is a triangle in the model space that satisfies , , and . A point for is called a comparison point if . Note, that for we need an additional condition on the perimeter of a triangle to guarantee existence of a comparison triangle, for the purposes of this post we will state the condition, but not elaborate on it in detail.
CAT(k)
So what’s a cat? It’s a gorgeous animal that many humans have as a pet. Many cats are fluffy, and so is mine. Also cats are silly and generally cool to have around. However, this post is not about these kinds of cats.
The term “CAT(k)” space was coined by Mikhail Gromov and consists of three initials honoring: Élie Cartan, Alexander D. Alexandrov, and Victor A. Toponogov. Now, we will define a CAT(k) space. Let be a metric space and let be a real number. Let be a geodesic triangle in with perimeter less than (the diameter of the space, this is the condition need to guarantee the existence of comparison triangle). Then we say that satisfies the CAT(k) inequality if for the corresponding comparison triangle and all with corresponding comparison points the inequality is satisfied. Thus, for a we will call a CAT(k) space if is geodesic and all its geodesic triangles satisfy the CAT(k) inequality. For , we relax definition by only requiring to be -geodesic and only requiring the inequality condition for triangles of perimeter bounded by .
Finally, we will call a space to be of curvature at most if it is locally a CAT(k) space. Hence, any space that is (locally) CAT(0) is a space on non-positive curvature (in the Alexandrov sense).
Intuitively speaking the triangles in a CAT(k) space have to be thinner than the ones in the corresponding model space. In particular, triangles in CAT(0) spaces are thinner than those in the regular Euclidean space (namely given two points on the sides of a triangle the distance between them in CAT(0) space is less than or equal to the distance between the corresponding comparison points in the Euclidean space). The illustration below shows three model spaces (sphere, plane, and hyperbolical paraboloid) and a cat drawn in each space. You can see that sphere cat is chubbier than the flat cat, which is chubbier than the hyperbolic cat, turns out triangles in these spaces are also of different chubbiness.
For the rest of the post we will focus specifically on the case of the CAT(0) spaces, as they present the most interest to us in the follow up posts.
An immediate, but important consequence of the definition of a CAT(k) space is that any CAT(0) space is uniquely geodesic. Note, that being uniquely geodesic also implies that a space is contractible (the converse does not hold, there are contractible spaces that are not uniquely geodesic, a simple example is the closed upper hemisphere which is clearly contractible, and also not uniquely geodesic for the same reason a sphere isn’t).
Next, we will briefly describe some interesting spaces which under certain conditions turn out to be CAT(0).
My CAT is a cubical complex!
Let be the unit interval, then we call the n-fold product the unit cube. We will let denote a point by convention. Since we will be mainly operating in dimensions, we will use the term “face” to describe any-dimensional face of the cube. Thus, the faces of are $\{0\}, \{1\}$ (the 0-dimensional faces), and (the 1-dimensional face). Analogously, for a faceis a subset which can be written as where each is a face of . The dimension of the face will be the sum of dimensions of , and we also note that a k-dimensional face will be isometric to .
Now, we are ready to define a cubical complex (Def. 7.32 from Bridson and Haefliger, 2013) in a similar vein to the definition of a simplicial complex (for those familiar with the latter construction). A cubical complex is the quotient of a disjoint union of cubes by an equivalence relation with the restrictions of the natural projection satisfying
for every the map is injective;
if then there is an isometry from a face onto a face such that .
While not precisely a cubical complex, the above rendition of a cat is reminiscent of one. The picture was taken from: https://www.boredpanda.com/i-made-animal-cube. All credit for creating those goes to the original author(s), which as far I found is: Aditya Aryanto.
Note, that in general a cubical complex needs not be a CAT(0) space. However, the following theorem of Gromov, 1987 gives the necessary and sufficient conditions for a cubical complex to be CAT(0).
Theorem 1. A cubical complex with the intrinsic Euclidean metric is CAT(0) if and only if is connected, simply connected, and for all natural : if three -cubes of share a common -cube and pairwise share common distinct -cubes, then they are contained within a -cube in .
A bit of perspective
In many cases the underlying geometry of the solution space can make or break our ability to solve problems efficiently. For example, many interesting discrete problems are NP-hard, and often do not even have good approximations (for a deeper dive Google “inapproximability results”, Khot, 2010 gives a great overview). In general, computing geodesics is a hard problem, but in certain spaces we can still efficiently compute (meaning in polynomial time) or approximate geodesics. CAT(0) cubical complexes are one particular class of “nice” spaces, meaning that geodesics can be computed or approximated well enough in polynomial time (Owen, and Provan, 2010, Hayashi, 2021). Implications of these algorithmic results mean that in certain phylogenetic (and robotics) problems we are able to efficiently compute distance between two points in the space, and reconstruct the optimal path between them (or at least do so up to error).
Code and data availability
All code used in this post is available in the following Jupyter notebook.
The next post in the series will take us back to the world of phylogeny, but this time armed with the knowledge about CAT(0) spaces. We will try to make sense of defining nice metrics on the space of phylogenetic trees with branch lengths, and learn a thing or two in the process. Essentially, the idea for the remainder of this stretch of the series will be to work our way through the paper of Gavryushkin and Drummond, 2016 on the space of ultrametric phylogenetic trees.
References
Bridson, Martin R., and André Haefliger. Metric spaces of non-positive curvature. Vol. 319. Springer Science & Business Media, 2013. https://doi.org/10.1007/978-3-662-12494-9
Gavryushkin, Alex, and Alexei J. Drummond. “The space of ultrametric phylogenetic trees.” Journal of theoretical biology 403 (2016): 197-208. arXiv:1410.3544
Hayashi, Koyo. “A polynomial time algorithm to compute geodesics in CAT (0) cubical complexes.” Discrete & Computational Geometry 65, no. 3 (2021): 636-654. arXiv:1710.09932
Khot, Subhash. “Inapproximability of NP-complete problems, discrete Fourier analysis, and geometry.” In Proceedings of the International Congress of Mathematicians 2010 (ICM 2010) (In 4 Volumes) Vol. I: Plenary Lectures and Ceremonies Vols. II–IV: Invited Lectures, pp. 2676-2697. 2010. https://cs.nyu.edu/~khot/papers/icm-khot.pdf
Owen, Megan, and J. Scott Provan. “A fast algorithm for computing geodesic distances in tree space.” IEEE/ACM Transactions on Computational Biology and Bioinformatics 8, no. 1 (2010): 2-13. https://doi.org/10.1109/TCBB.2010.3
Often in biology applications we are interested not only in inferring the optimal tree under some evolutionary model, but also in comparing two different trees for the same set of taxa (or even two different trees for two overlapping but not necessarily identical sets of taxa), it is natural to ask a question about the the dissimilarity measures between trees. In general, the question of defining a metric over the space of phylogenetic trees is quite rich and interesting, and we will explore this in more detail over several posts. However, it is useful to start with the simplest case in which we compare two unrooted trees without branch lengths over a fixed set of taxa. While multiple methods have been proposed to address this question, the metric proposed by Robinson and Foulds, 1981 remains a popular choice in practice (the paper has more than 2.6k citations according to Google Scholar), and more importantly for us serves as a great starting point for the exploration of the metrics on tree spaces.
Before we dive into the details of Robinson-Foulds metric it might be useful to briefly review some of the general metric space concepts and definitions.
Review: Metric spaces
A metric space is an ordered pair where is a set and is a function satisfying
for any . Recall from the previous post that in the case when the third condition (called triangle inequality) is replaced by a stricter condition we obtain an ultrametric space.
Note that given any set we can create a simple metric on it by setting if and otherwise. This metric is called the discrete metric (and it is trivial to check that discrete metric is also an ultrametric). Discrete metric provides us with an important realization that any set can be turned into a metric space. However, it is clear that discrete metric is not useful in a practical comparison scenario.
We also note that a metric on a set gives rise to topology, so any metric space is also a topological space. The base for topology on a metric space is defined by the open balls .
In case when the underlying set is finite, we call the resulting metric space a finite metric space. Note that if we consider trees without branch lengths then the resulting set is finite, and hence the corresponding metric space is a finite metric space. This is not the case when we consider the branch lengths, but we will leave the discussion of the geometry of phylogenetic trees with branch lengths to later posts.
Robinson-Foulds metric
Given a set of taxa let us define the set of all unrooted (not necessarily bifurcating) trees with the property that all nodes of degree 1 or 2 are labeled by pairwise disjoint non-empty subsets of as . Below are a few examples of trees in for the set of taxa . Unlabeled nodes are considered to have as their label.
Conversely neither of the trees shown below are in , since they violate the labeling properties.
Note, that any unrooted phylogenetic tree on will be an element of , but not all trees in are fully resolved unrooted phylogenies.
Now, we will define two operations on named contraction (denoted by ) and decontraction (denoted ).
Contraction will consist of an edge contraction together with the union operation on the corresponding node label sets. More precisely given a tree with the labeling defined by and an edge we will define the contraction by considering and adding a vertex with and edges . Thus, we have . The figure below illustrates an example of subsequent contractions on tree in for .
Decontraction will consist of an “inverse” operation in which we will split a node by creating an edge. Note that the caveat here is that such an operation depends not only on the choice of the node to split, but also on the choice of a label set and incident edges set split. Hence, the use of notation is misleading since applying a decontraction after a contraction does not necessarily result in the original tree. Hence, to define a decontraction precisely we will define it as a function . Let where and . Let where and . Now, we can construct the tree and add two vertices to it alongside the edge and redistribute the edges previously incident on . Formally, with the labeling function defined as and for the new nodes. The figure below illustrates two of the possible ways a decontraction can be applied to tree in .
Now, we can finally define the Robinson-Foulds metric on the space of trees . We let denote the minimum number of applications of the and operations required to transform into (whenever we say that two trees are the same we mean that there exists a label preserving isomorphism).
First, let’s make sure that the distance function is well-defined. We note that any operation can be reversed via an appropriately chosen operation. Furthermore, it is clear that for any tree within the operations we will obtain a tree consisting of a single node with the label (representing all taxa) and no edges. Thus, for any two trees it follows that we can transform both to the tree with one node, and then apply appropriate inverse transformations, implying that there is a finite (and in fact bounded above by ) sequence of operations that transforms into (and vice versa). We will not prove that is indeed a metric, but the proofs of each of the properties are easy.
Robinson-Foulds metric: properties and caveats
Robinson-Foulds (RF) metric comes with several nice properties which made it ubiquitous in the world of phylogenetic analyses: (1) it can be efficiently computed (linear time in number of taxa via Day, 1985 or approximate solution in sub-linear time), and (2) it has intuitive interpretation as the number of distinct splits between the two trees (for more details see Felsenstein, 2004). However, RF metric also suffers from several issues. First, the placement of a single taxon can easily produce maximum RF distance between two otherwise identical trees. The figure below illustrates this example for two unrooted trees with 5 taxa.
Furthermore, the problem of large RF distances is a general issue with this metric. For example, we have generated 945 rooted trees on 6 taxa, and computed pairwise distance between distinct pairs of trees. The resulting histogram of normalized RF distances is shown below.
As we can see more than a third of all distances achieved the maximum possible value. Put differently, if we view the discrete metric as the least informative (everything is distance 1 away from any other point) then RF metric is only slightly better than the discrete one. This obviously presents us with a problem when we want our comparisons to provide a relative ranking of similarity with respect to a reference tree or when we want to use the distances as a tool for comparison between multiple sets of data.
There are a number of refinements of the RF metric which attempt to overcome some of the issues we pointed out above. Of particular interest is recent work of Smith, 2020 in which the author proposes an information theoretic generalization of the RF metric. Key idea in this paper is to use the information content of each split in order to weight its impact on the measured distance. Furthermore, the paper provides a set of comparisons with other generalizations of the RF metric, and as such can serve as a good overview of the topic for readers curious to learn more about the current state of the field.
Data and code availability
All code used in this post is available as a Jupyter notebook in the attached archive.
Robinson-Foulds metric serves as an important example at the start of the journey into tree geometry and comparative analyses. However, since it is purely focused on the topology of the tree it lacks expressiveness required to compare phylogenetic trees with branch lengths. Thus, we need different methods in order to compare those. Furthermore, the arising geometries (spoiler alert: there is more than one) are quite interesting in themselves. Thus, we will likely return to the topic of tree distances later on in the series.
References
Day, William HE. “Optimal algorithms for comparing trees with labeled leaves.” Journal of classification 2, no. 1 (1985): 7-28. https://doi.org/10.1007/BF01908061
Robinson, David F., and Leslie R. Foulds. “Comparison of phylogenetic trees.” Mathematical biosciences 53, no. 1-2 (1981): 131-147. https://doi.org/10.1016/0025-5564(81)90043-2
Smith, Martin R. “Information theoretic generalized Robinson–Foulds metrics for comparing phylogenetic trees.” Bioinformatics 36, no. 20 (2020): 5007-5013. https://doi.org/10.1093/bioinformatics/btaa614
In an effort to learn more about phylogenetics and practice my writing skills I am starting a mini-series of phylogenetic vignettes in which I will explore different concepts in phylogenetic analysis and their implementations. This series is intended as a partial account of my journey in learning some of the topics in phylogenetics. The resulting posts are not intended to be complete explorations of the corresponding questions, and will be presented at the current level of author’s understanding which can be incomplete. The intended audience for these vignettes are undergraduate and graduate students with solid understanding of basic concepts in discrete mathematics, as well as some knowledge of analysis and probability. I will assume no deep knowledge of evolutionary biology, in part due to my own background lacking strong biology foundation.
a finite set of taxa (for non-bio folks: a set of labels)
a unrooted phylogenetic tree, i.e. a undirected connected graph with no cycles and in which together with a leaf labeling that assigns exactly one taxon to each leaf and none to any internal node
a weight assignment for a given phylogenetic tree , i.e. a map (in practice we are interested only in the case )
a pairwise distance matrix for the set of genomic sequences corresponding to the set of taxa
We will also use the term molecular clock to refer to the situations in which the rate of mutation is fixed per unit of time. Thus the molecular clock hypothesis is a useful assumption for inference of trees for the sequences that were collected at the same point in time, hence we can assume in that scenario that in the rooted phylogenetic tree distance from root to every leaf is constant.
Distance based tree inference
In general, phylogenetic tree inference problem consists of inferring an optimal rooted or unrooted phylogenetic tree given a set of sequences representing taxa . Here by optimal we mean optimization with respect to some evolutionary model over the full parameter space defined by topologies of and weights .
The exact criterion with respect to which the optimization is done varies between various formulations of the problem. However, one thing that is common to all formulations is the prohibitively large size of the search space. A simple calculation shows that the number of bifurcating trees (i.e. every non-leaf node has exactly two children or alternatively every non-leaf has degree 3) on taxa is .
In the case of distance based phylogenetic tree inference our input is the pairwise distance matrix . We will explore different options for measuring distance between input sequences in a later section.
Least squares
We can start with an attempt at defining an optimization goal for our tree inference problem given the input distance matrix . Intuitively the goal here is to find a tree that best approximates (explains) the observed distances. More formally, given a phylogenetic tree with edge weights we can define the tree-induced metric on the set of taxa as , where we use the notation to denote the path between leaves of the tree (in a tree any two nodes are connected by a single path). Thus, we want to minimize the error under some norm. One possible choice of the norm is the Frobenius norm, which in turn corresponds to unweighted least squares tree inference. As noted in Felsenstein, 2004 the least squares formulation can be weighted, i.e. the function to minimize becomes with the choice proposed by Cavalli-Sforza and Edwards, 1967, by Fitch and Margoliash, 1967, and by Beyer et al., 1974.
While for a fixed tree topology the problem boils down to least squares optimization, the general problem of tree inference under this model requires searching over the large space of trees. Thus, while the formulation behind this method is sound and appealing, in practice we tend to rely on heuristics when searching the tree space.
Instead of fully exploring least squares method and its derivatives and search heuristics we will now pivot to a different approach in which instead of explicit optimization under a given metric we will instead focus on direct clustering goal.
UPGMA
The first approach we will explore is called Unweighted Pair Group Method with Arithmetic Mean (UPGMA). For those familiar with clustering methods, and hierarchical clustering in particular, this name might ring a bell. It appears that the name UPGMA mostly stuck with the phylogenetics community, and in the general clustering world this approach is referred to as agglomerative (i.e. bottom-up) clustering with average linkage (scikit-learn reference, scipy reference).
Implementing UPGMA naively is straightforward and we will illustrate it in the following code snippet with the distance matrix data coming from Felsenstein, 2004 (pp. 162-166).
Note that the above implementation doesn’t keep track of the branch lengths when providing the output and as a result our tree has the correct topology but misses the edge weight information. Alternatively we can simply use Scipy hierarchical clustering module and achieve the same result including the branch lengths in fewer lines of code.
One thing that becomes apparent from the second plot is that the resulting tree is an ultrametric tree.
In general we call a metric space and ultrametric space if in addition to the standard properties the metric satisfies the strong triangle inequality for all . Ultrametric spaces are somewhat bizarre looking since every triangle in such space is isosceles. Furthermore, every point inside an open ball of positive radius is its center, and all open balls of positive radius are clopen. However, ultrametric spaces arise naturally in several settings with the one of interest to us being phylogenetic trees under the molecular clock hypothesis. Such trees are commonly referred to as ultrametric trees.
Now, one of the interesting properties of the UPGMA algorithm is that the tree constructed by this method will always be ultrametric. However, this also implies that if the original distance matrix does not induce an ultrametric on then the tree produced by the UPGMA algorithm will have a non-zero error . For example, consider the following sample distance matrix given as an example in Huson, Rupp, and Scornavacca, 2010 (p. 53):
A
B
C
D
E
A
0
2
7
5
4
B
2
0
7
5
4
C
7
7
0
4
5
D
5
5
4
0
3
E
4
4
5
3
0
The inferred UPGMA tree will look as following
Due to the metric induced by the tree the distances between C and D, and C and E, will be equal in while we know from the original distance matrix that the distance from C to D is supposed to be larger. Thus, UPGMA model is not broadly applicable since it does require the original distance matrix to satisfy the ultrametric condition. Conversely, Huson, Rupp, and Scornavacca, 2010Lemma 3.13.2 shows that if the distance matrix is ultrametric then the tree inferred by UPGMA satisfies .
In terms of the computational complexity UPGMA requires time, which is a significant speed up when compared to brute-force search over the tree space.
Felsenstein, 2004 notes that in some scenarios, for example for closely related species, where the molecular clock is a reasonable starting assumption the method beahves well. However, in the general setting where we cannot claim a fixed rate for the whole tree UPGMA approach becomes problematic.
Neighbor-joining
Neighbor-joining is an alternative method for inferring a phylogenetic tree from the distance matrix . Originally it was proposed by Saitou and Nei, 1987 and has since been implemented in several libraries and software packages. We will use the implementation provided in scikit-bio (in case if you get a conflict with Scipy, try downgrading Scipy to version 1.5.2), and skip discussing the details of the algorithm in this post.
Motivation for considering neighbor-joining is that it doesn’t require the underlying distance matrix to be ultrametric in order to faithfully infer a phylogenetic tree. However, in order to guarantee that the algorithm will infer a tree with we still require that the distance matrix is “tree-like”. This vague term can be made precise via the Definition 3.12.4 from Huson, Rupp, and Scornavacca, 2010 (p. 51) also referred to as the four-point condition: a distance matrix is said to satisfy the four-point condition if for every quadruple of taxa we have . In the case when the distance matrix satisfies the above condition we can guarantee that for the inferred tree . The proof of the above claim can be found in the original paper.
Neighbor-joining has the same computational complexity as UPGMA, and both methods are agglomerative clustering approaches that differ in the distance and update rules. One interesting aspect of the neighbor joining method is that it has been shown to greedily optimize balanced minimum evolution metric on the tree space (Gascuel and Steel, 2006). We will not explore the details of the balanced minimum evolution model at this point, but we might return to the topic in later post.
Exploring sequence distance metrics
So far we only considered examples in which we had a predefined distance matrix provided to us from the beginning. In this section we will explore a few choices for constructing given a set of genomic sequences and compare how different distance based inference methods fair in these scenarios.
Jukes-Cantor model and distance
The simplest substitution model used in phylogenetics is the Jukes-Cantor model. Under this model we assume that the substitution rate is fixed and equal and the base frequencies are all equal to 1/4. Under these assumptions it is easy to derive the correspondence between the expected number of substitutions (i.e. evolutionary distance) and the fraction of distinct bases in the alignment (i.e. normalized Hamming distance). Thus, given two aligned sequences we will refer to the quantity , where is the normalized Hamming distance, as the Jukes-Cantor distance between and .
Mash distance
MinHash sketching is an effective computational technique for estimating similarity of two sets of data. Due to its computational efficiency MinHash has found its into many data intensive areas of computer science, including bioinformatics. In particular, due to the computational efficiency it’s common that we view sequences as sets (or multi-sets) of k-mers (substrings of length k, think k-grams in NLP), and hence we can compare the (Jaccard) similarity of two sequences via MinHash on the respective k-mers. However, upon closer inspection it is clear that Jaccard distance is not a good proxy for the evolutionary distance (similarly how normalized Hamming distance is not exactly the same thing as Jukes-Cantor distance). Thus, we want to compute an appropriate adjustment that turns our estimated Jaccard index into an approximate evolutionary metric. One possible approach is given by Ondov et al., 2006, Eq. 4 which defines Mash distance between two sequences (or sets of sequences) as where is the length of the k-mer and is the estimated Jaccard index.
Comparing UPGMA and Neighbor-joining for JC distance
We will begin with a set of 5 sequences each consisting of 70 nucleotides and containing 5 single nucleotide changes with respect to the first 70 nucleotides of the N gene of the reference genome for SARS-CoV-2.
We will label the sequences in this set A, B, C, D, and E in top to bottom order. We get the following matrix of Jukes-Cantor distances for this set:
A
B
C
D
E
A
0
0.158
0.158
0.158
0.124
B
0.158
0
0.044
0.075
0.124
C
0.158
0.044
0
0.107
0.107
D
0.158
0.075
0.107
0
0.059
E
0.124
0.124
0.107
0.059
0
It is easy to check that this matrix is not additive, and hence is also not an ultrametric. Thus, we do not expect either of the algorithms to yield an optimal tree. Nevertheless we would like to compare the two topologies we get. First we will consider the UPGMA tree:
We can see that the sequences A ends up acting as an outgroup, which is not surprising if we return and examine the mutation content within the sequence. Sequences B and C, and D and E form two clades in the UPGMA tree. Examining the alignment by eye it appears as a plausible grouping. However, we note that the distance between sequences B and D is estimated to be smaller than that between B and E, while the inferred tree does not capture this difference.
Next, we will consider the tree obtained from the neighbor-joining method.
BCDEA
(function()
{
var modules={};
})();
Similarly to the UPGMA tree the sequences B and C end up in the same clade in the neighbor-joining tree. Analogously sequence A appears to form an outgroup. The key difference in this case is the way in which sequences D and E are placed onto the tree. However, neighbor-joining tree also fails to capture some of the differences in the distances, for example sequence E is equidistant from B and C in the tree.
Comparing UPGMA and Neighbor-joining for Mash distance
We will start out with the same sequence setup as in the previous run, except instead of the Jukes-Cantor distances we will use Mash distances this time. We are setting the k-mer size to 7 for this scenario, and we obtain the following distance matrix:
A
B
C
D
E
A
0
0.158
0.158
0.144
0.121
B
0.158
0
0.027
0.057
0.079
C
0.158
0.027
0
0.071
0.071
D
0.144
0.057
0.071
0
0.034
E
0.121
0.079
0.071
0.034
0
Unsurprisingly this matrix is not additive either, and the distance values tend to correlate with the Jukes-Cantor distances. Thus, as expected both inferred trees have the same topologies as in the previous section, albeit slightly different branch lengths.
BCDEA
(function()
{
var modules={};
})();
Now, we will try to perform a similar comparison but with a different underlying dataset. We downloaded 454 complete SARS-CoV-2 genomes collected between August 1st, 2022 and August 15th, 2022 from NCBI Virus database. We then computed the Mash distances between all these sequences using 11-, 17-, 23,-, and 31-mers. Finally for each distance matrix we inferred the phylogenetic tree using UPGMA and neighbor-joining methods.
In the above dendrogram we truncated subtrees after a fixed depth level for the ease of visualization, the labels when present indicate a leaf node with the corresponding PANGO lineage assigned to it. One thing that we immediately notice in this case is that BA.2, BA.5 (and BA.4, labels not shown) form a clade, and B.1.617.2, AY.* form a clade. This matches the expected behavior since, the first clade corresponds to the BA.2-related sublineages of Omicron variant, and the second clade corresponds to Delta variant. What is slightly surprising is that BA.1 (also a sublineage of Omicron) ends up in a clade with B.1.616 and B.1 lineages, rather than next to BA.2. We also inferred a neighbor-joining based tree and adopted leaf coloring scheme similar to the one in UPGMA.
(function()
{
var modules={};
})();
In this tree the deeper shade of orange represents BA.5 and its sublineages, while lighter shade of orange corresponds to BA.2 and BA.4 sublineages of Omicron. Green is used for Delta, and red for everything else. One thing that becomes more apparent with this tree is that there is subclade consisting predominantly of BA.5 sequences that is at a considerable distance from the rest of the sequences in the tree.
Another interesting observation we get from this experiment is that the k-mer size used did not influence the resulting tree shape drastically. While there are some minor changes occurring as we increase the size of k, the main clades centered around BA.5 and Delta emerge in all inferred trees. This suggests a practical observation that in the case of a large amount of closely related sequences, we can apply very approximate methods (since we stacked Mash distance with UPGMA we effectively “approximated” twice) and still capture some underlying structure in the data.
Data and code availability
In addition to the two Gists shared above you can also find the Jupyter Notebook with the code used to generate figures for this post, as well as sequence data and precomputed Mash distances in the archive attached below.
We very briefly touched upon some of the distance-based phylogenetic inference methods and checked out several trees inferred from two evolutionary distance metrics. However, we haven’t rigorously defined how exactly two tree topologies and two phylogenetic trees can be compared. We will dive into that topic over the course of the next few posts.
References
Beyer, William A., Myron L. Stein, Temple F. Smith, and Stanislaw M. Ulam. “A molecular sequence metric and evolutionary trees.” Mathematical Biosciences 19, no. 1-2 (1974): 9-25. https://doi.org/10.1016/0025-5564(74)90028-5
Gascuel, Olivier, and Mike Steel. “Neighbor-joining revealed.” Molecular biology and evolution 23, no. 11 (2006): 1997-2000. https://doi.org/10.1093/molbev/msl072
Haeckel, E. H. P. A. (1866).Generelle Morphologie der Organismen : allgemeine Grundzüge der organischen Formen-Wissenschaft, mechanisch begründet durch die von C. Darwin reformirte Decendenz-Theorie. Berlin. [Featured image]
Huson, Daniel H., Regula Rupp, and Celine Scornavacca. Phylogenetic networks: concepts, algorithms and applications. Cambridge University Press, 2010.
Ondov, Brian D., Todd J. Treangen, Páll Melsted, Adam B. Mallonee, Nicholas H. Bergman, Sergey Koren, and Adam M. Phillippy. “Mash: fast genome and metagenome distance estimation using MinHash.” Genome biology 17, no. 1 (2016): 1-14. https://doi.org/10.1186/s13059-016-0997-x
Saitou, Naruya, and Masatoshi Nei. “The neighbor-joining method: a new method for reconstructing phylogenetic trees.” Molecular biology and evolution 4, no. 4 (1987): 406-425. https://doi.org/10.1093/oxfordjournals.molbev.a040454





![[n]=\{1, 2, 3, ..., n\}](https://s0.wp.com/latex.php?latex=%5Bn%5D%3D%5C%7B1%2C+2%2C+3%2C+...%2C+n%5C%7D&bg=fafafa&fg=6c6c8c&s=0&c=20201002)
![f:[n]\to[n]](https://s0.wp.com/latex.php?latex=f%3A%5Bn%5D%5Cto%5Bn%5D&bg=fafafa&fg=6c6c8c&s=0&c=20201002)
![\mathcal{F}_n=\{f|f:[n]\to[n]\}](https://s0.wp.com/latex.php?latex=%5Cmathcal%7BF%7D_n%3D%5C%7Bf%7Cf%3A%5Bn%5D%5Cto%5Bn%5D%5C%7D&bg=fafafa&fg=6c6c8c&s=0&c=20201002)

![i\in[n]](https://s0.wp.com/latex.php?latex=i%5Cin%5Bn%5D&bg=fafafa&fg=6c6c8c&s=0&c=20201002)


![j\in[n]](https://s0.wp.com/latex.php?latex=j%5Cin%5Bn%5D&bg=fafafa&fg=6c6c8c&s=0&c=20201002)


![[n]](https://s0.wp.com/latex.php?latex=%5Bn%5D&bg=fafafa&fg=6c6c8c&s=0&c=20201002)



































![\sigma:[n]\to[n]](https://s0.wp.com/latex.php?latex=%5Csigma%3A%5Bn%5D%5Cto%5Bn%5D&bg=fafafa&fg=6c6c8c&s=0&c=20201002)





![\mathbb{E}[X]=\sum_{\sigma}\mathbb{E}[X_\sigma]=\sum_{\sigma}Pr(X_\sigma =1)](https://s0.wp.com/latex.php?latex=%5Cmathbb%7BE%7D%5BX%5D%3D%5Csum_%7B%5Csigma%7D%5Cmathbb%7BE%7D%5BX_%5Csigma%5D%3D%5Csum_%7B%5Csigma%7DPr%28X_%5Csigma+%3D1%29&bg=fafafa&fg=6c6c8c&s=0&c=20201002)
![\mathbb{E}[X]=(n-1)!/2^n](https://s0.wp.com/latex.php?latex=%5Cmathbb%7BE%7D%5BX%5D%3D%28n-1%29%21%2F2%5En&bg=fafafa&fg=6c6c8c&s=0&c=20201002)

































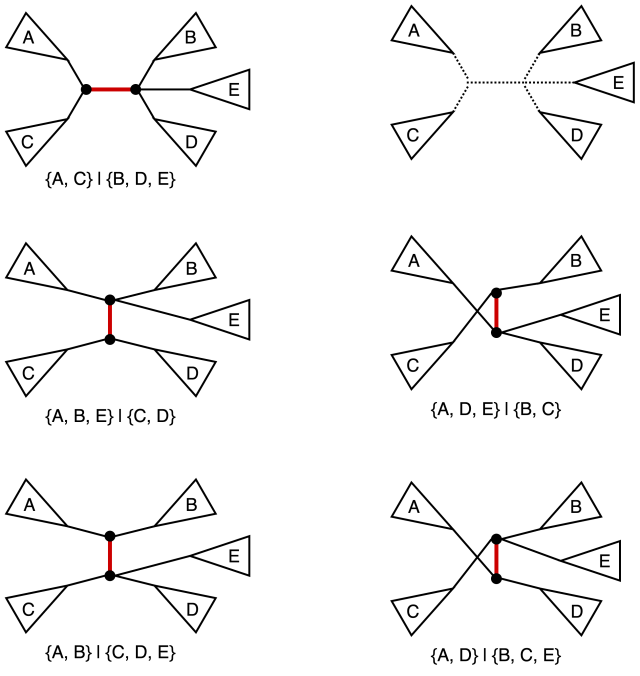



 .
.  between points
between points  and
and  . Choose a set of points
. Choose a set of points  such that
such that  . Note that it follows from the definition of a local geodesic that
. Note that it follows from the definition of a local geodesic that  is a geodesic. Now, assume by induction that
is a geodesic. Now, assume by induction that  is a geodesic for all $j\leq i$. Hence, we have
is a geodesic for all $j\leq i$. Hence, we have  by the induction hypothesis and
by the induction hypothesis and  as noted above. Consider a corresponding model triangle
as noted above. Consider a corresponding model triangle  in Euclidean space (if you forgot what a model triangle is check:
in Euclidean space (if you forgot what a model triangle is check:  be a point on
be a point on  such that
such that  . Since we are working in a CAT(0) space it follows that
. Since we are working in a CAT(0) space it follows that  . On the other hand, by induction hypothesis we have
. On the other hand, by induction hypothesis we have  . Combining this result with triangle inequality gives us
. Combining this result with triangle inequality gives us  , and hence
, and hence  and therefore
and therefore  . From here we can conclude that
. From here we can conclude that  is a geodesic and hence $\Gamma_\varepsilon$ is a geodesic.
is a geodesic and hence $\Gamma_\varepsilon$ is a geodesic. between which we are computing geodesics have distinct edges. We will also call two sets of edges
between which we are computing geodesics have distinct edges. We will also call two sets of edges  and
and  compatible if all pairs of splits associated with these edges are compatible, or equivalently if the union
compatible if all pairs of splits associated with these edges are compatible, or equivalently if the union  defines a tree. In other words, compatible sets of edges produce “intermediate” trees between
defines a tree. In other words, compatible sets of edges produce “intermediate” trees between  and
and  . Now, for a pair of partitions
. Now, for a pair of partitions  ,
,  of
of  and
and  , respectively, we can define an associated path space given that the following property holds: for every
, respectively, we can define an associated path space given that the following property holds: for every  ,
,  and
and  are compatible. To construct the path space consider the collection of orthants
are compatible. To construct the path space consider the collection of orthants  given by
given by  , where
, where  is the mapping of a tree to the corresponding orthant. We will call the corresponding partition pair
is the mapping of a tree to the corresponding orthant. We will call the corresponding partition pair  the support, and the shortest path between
the support, and the shortest path between  the path space geodesic. These definitions make the geodesic in the tree space a more concrete and constructable object, which of course is important for any algorithmic approach. Furthermore, the following theorem from
the path space geodesic. These definitions make the geodesic in the tree space a more concrete and constructable object, which of course is important for any algorithmic approach. Furthermore, the following theorem from  .
. be a geodesic between
be a geodesic between  latex \mathcal{B}=\{B_1,…,B_k\} of
latex \mathcal{B}=\{B_1,…,B_k\} of  .
. -path
-path  in the support there is no nontrivial partition
in the support there is no nontrivial partition  ,
,  such that
such that  is compatible with
is compatible with  and
and  .
. and the corresponding path is given by uniform contraction of all edges which results in star tree and then a follow up uniform decontraction. Now, the algorithm will proceed iteratively at each step checking whether the condition in Theorem 3 is satisfied and if not proposing a new support and a new proper path.
and the corresponding path is given by uniform contraction of all edges which results in star tree and then a follow up uniform decontraction. Now, the algorithm will proceed iteratively at each step checking whether the condition in Theorem 3 is satisfied and if not proposing a new support and a new proper path.  latex A\subseteq E(T), B\subseteq E(T^\prime)$ as a bipartite graph with vertices given by
latex A\subseteq E(T), B\subseteq E(T^\prime)$ as a bipartite graph with vertices given by  where the splits associated with
where the splits associated with  and
and  are incompatible. It is easy to see that for any
are incompatible. It is easy to see that for any  compatibility of
compatibility of  and
and  is equivalent to
is equivalent to  ,
,  such that the vertex set
such that the vertex set  is independent in
is independent in  and
and  satisfies the above condition.
satisfies the above condition. . Further analysis of the algorithm steps yields that the total runtime to find the geodesic is
. Further analysis of the algorithm steps yields that the total runtime to find the geodesic is  . For the exact details of the proof of correctness of the algorithm and the runtime I suggest reading the original manuscript by
. For the exact details of the proof of correctness of the algorithm and the runtime I suggest reading the original manuscript by  into some metric space
into some metric space  . Thus, by associating every tree in the tree space with some point in the chosen metric space, we induce a pseudometric on tree space. However, such embeddings/parameterizations are not guaranteed to be “nice” with respect to the questions we aim to answer. Namely, not all embeddings are injective (multiple distinct trees can end up being mapped to the same point in
. Thus, by associating every tree in the tree space with some point in the chosen metric space, we induce a pseudometric on tree space. However, such embeddings/parameterizations are not guaranteed to be “nice” with respect to the questions we aim to answer. Namely, not all embeddings are injective (multiple distinct trees can end up being mapped to the same point in  , hence in general we induce a pseudometric rather than a metric via embedding) although in the case of
, hence in general we induce a pseudometric rather than a metric via embedding) although in the case of  that would enable fruitful analyses. From now onwards (unless otherwise specified) we will assume that the embedding
that would enable fruitful analyses. From now onwards (unless otherwise specified) we will assume that the embedding  is injective.
is injective.  is path-connected in
is path-connected in  , where $\tau_i$ is the time difference between the i-th and i+1-th nodes. The picture below taken from
, where $\tau_i$ is the time difference between the i-th and i+1-th nodes. The picture below taken from 
 that embeds the space of ultrametric phylogenetic trees into a disjoint union of
that embeds the space of ultrametric phylogenetic trees into a disjoint union of  (
( ). We will impose an upper bound on all orthants to turn our construction into a cubical complex for the ease of the exposition. However, the construction of 𝛕-space with orthants will still yield a CAT(0) space with the key properties and desiderata (D1)-(D5′) preserved.
). We will impose an upper bound on all orthants to turn our construction into a cubical complex for the ease of the exposition. However, the construction of 𝛕-space with orthants will still yield a CAT(0) space with the key properties and desiderata (D1)-(D5′) preserved.
 face. Since the resulting space is a cubical complex, we can naturally consider the Euclidean metric within each cube with paths for joining points in different cubes being the sums of the respective within cube paths.
face. Since the resulting space is a cubical complex, we can naturally consider the Euclidean metric within each cube with paths for joining points in different cubes being the sums of the respective within cube paths. , then the resulting facet is only contained in the cube of corresponding rooted topology. If setting
, then the resulting facet is only contained in the cube of corresponding rooted topology. If setting  does not result in a multifurcating topology then the corresponding facet will be shared by exactly two cubes (see example in Figure 2 and/or Figure 2 of
does not result in a multifurcating topology then the corresponding facet will be shared by exactly two cubes (see example in Figure 2 and/or Figure 2 of  with the intrinsic Euclidean metric is CAT(0) if and only if
with the intrinsic Euclidean metric is CAT(0) if and only if  : if three
: if three  -cubes of
-cubes of  -cubes, then they are contained within a
-cubes, then they are contained within a  -cube in
-cube in  . Hence, there exists
. Hence, there exists  s.t. it is greater than zero in both of the first two cubes, and is zero in their shared (k+1)-cube. Now, if the first and third cube share a (k+1)-cube then it follows that their i and r coordinates both have to be zero in the shared cube, implying that the s coordinate has to be resolved in the same way between the first and third cube. However, the same exact argument can be repeated for the second and third cube implying that the first and second cubes had to be indetical.
s.t. it is greater than zero in both of the first two cubes, and is zero in their shared (k+1)-cube. Now, if the first and third cube share a (k+1)-cube then it follows that their i and r coordinates both have to be zero in the shared cube, implying that the s coordinate has to be resolved in the same way between the first and third cube. However, the same exact argument can be repeated for the second and third cube implying that the first and second cubes had to be indetical.
 a geodesic from
a geodesic from  is the map
is the map ![c:[0, l]\to X](https://s0.wp.com/latex.php?latex=c%3A%5B0%2C+l%5D%5Cto+X&bg=fafafa&fg=6c6c8c&s=0&c=20201002) s.t.
s.t.  and
and  . We will call a metric space a geodesic space if for any
. We will call a metric space a geodesic space if for any  there exists a geodesic from
there exists a geodesic from  we have the geodesic given by
we have the geodesic given by  . On a sphere geodesic between two points
. On a sphere geodesic between two points  is the arc segment obtained by intersecting a plane through
is the arc segment obtained by intersecting a plane through 
 is a set of tree points
is a set of tree points  called vertices and a choice of three geodesic segments
called vertices and a choice of three geodesic segments ![[p, q], [q, r], [r, p]](https://s0.wp.com/latex.php?latex=%5Bp%2C+q%5D%2C+%5Bq%2C+r%5D%2C+%5Br%2C+p%5D&bg=fafafa&fg=6c6c8c&s=0&c=20201002) joining them called sides, we will denote such a triangle
joining them called sides, we will denote such a triangle ![\Delta([p, q], [q, r], [r, p])](https://s0.wp.com/latex.php?latex=%5CDelta%28%5Bp%2C+q%5D%2C+%5Bq%2C+r%5D%2C+%5Br%2C+p%5D%29&bg=fafafa&fg=6c6c8c&s=0&c=20201002) or more briefly
or more briefly  . Note, that the later notation is not precise, as in the case of a non-uniquely geodesic space there is not necessarily a unique choice of a geodesic between two vertices. Finally, we will write
. Note, that the later notation is not precise, as in the case of a non-uniquely geodesic space there is not necessarily a unique choice of a geodesic between two vertices. Finally, we will write  to indicate that
to indicate that ![[p, q]\cup [q, r]\cup [r, p]](https://s0.wp.com/latex.php?latex=%5Bp%2C+q%5D%5Ccup+%5Bq%2C+r%5D%5Ccup+%5Br%2C+p%5D&bg=fafafa&fg=6c6c8c&s=0&c=20201002) .
.  Euclidean n-dimensional space,
Euclidean n-dimensional space,  the n-sphere, and
the n-sphere, and  hyperbolic n-space. We already defined geodesics in the case of
hyperbolic n-space. We already defined geodesics in the case of  . For the n-sphere we will define the metric via cosine distance
. For the n-sphere we will define the metric via cosine distance  where the inner product is in the corresponding embedding
where the inner product is in the corresponding embedding  and the corresponding geodesics are given by minimal great arcs (for full definition and description see
and the corresponding geodesics are given by minimal great arcs (for full definition and description see  which consists of
which consists of  with bilinear form
with bilinear form  and defining
and defining  . The distance on this space will be given by
. The distance on this space will be given by  (similarly to the sphere case, see
(similarly to the sphere case, see  is a triangle in the model space
is a triangle in the model space  that satisfies
that satisfies  ,
,  , and
, and  . A point
. A point ![\overline{x}\in[\overline{p}, \overline{q}]](https://s0.wp.com/latex.php?latex=%5Coverline%7Bx%7D%5Cin%5B%5Coverline%7Bp%7D%2C+%5Coverline%7Bq%7D%5D&bg=fafafa&fg=6c6c8c&s=0&c=20201002) for
for ![x\in[p, q]](https://s0.wp.com/latex.php?latex=x%5Cin%5Bp%2C+q%5D&bg=fafafa&fg=6c6c8c&s=0&c=20201002) is called a comparison point if
is called a comparison point if  . Note, that for
. Note, that for  we need an additional condition on the perimeter of a triangle to guarantee existence of a comparison triangle, for the purposes of this post we will state the condition, but not elaborate on it in detail.
we need an additional condition on the perimeter of a triangle to guarantee existence of a comparison triangle, for the purposes of this post we will state the condition, but not elaborate on it in detail.  (the diameter of the
(the diameter of the  satisfies the CAT(k) inequality if for the corresponding comparison triangle
satisfies the CAT(k) inequality if for the corresponding comparison triangle  and all
and all  with corresponding comparison points
with corresponding comparison points  the inequality
the inequality  is satisfied. Thus, for a
is satisfied. Thus, for a  we will call
we will call  -geodesic and only requiring the inequality condition for triangles of perimeter bounded by
-geodesic and only requiring the inequality condition for triangles of perimeter bounded by 
![I=[0, 1]](https://s0.wp.com/latex.php?latex=I%3D%5B0%2C+1%5D&bg=fafafa&fg=6c6c8c&s=0&c=20201002) be the unit interval, then we call the n-fold product
be the unit interval, then we call the n-fold product  the unit cube. We will let
the unit cube. We will let  denote a point by convention. Since we will be mainly operating in
denote a point by convention. Since we will be mainly operating in ![[0, 1]](https://s0.wp.com/latex.php?latex=%5B0%2C+1%5D&bg=fafafa&fg=6c6c8c&s=0&c=20201002) (the 1-dimensional face). Analogously, for
(the 1-dimensional face). Analogously, for  which can be written as
which can be written as  where each
where each  is a face of
is a face of  . The dimension of the face will be the sum of dimensions of
. The dimension of the face will be the sum of dimensions of  .
.  by an equivalence relation
by an equivalence relation  with the restrictions
with the restrictions  of the natural projection
of the natural projection  satisfying
satisfying the map
the map  is injective;
is injective; then there is an isometry
then there is an isometry  from a face
from a face  onto a face
onto a face  such that
such that  .
.
 error).
error). is a function
is a function  satisfying
satisfying


 . Recall from the previous post that in the case when the third condition (called triangle inequality) is replaced by a stricter condition
. Recall from the previous post that in the case when the third condition (called triangle inequality) is replaced by a stricter condition  we obtain an ultrametric space.
we obtain an ultrametric space.  if
if  and
and  otherwise. This metric is called the discrete metric (and it is trivial to check that discrete metric is also an ultrametric). Discrete metric provides us with an important realization that any set can be turned into a metric space. However, it is clear that discrete metric is not useful in a practical comparison scenario.
otherwise. This metric is called the discrete metric (and it is trivial to check that discrete metric is also an ultrametric). Discrete metric provides us with an important realization that any set can be turned into a metric space. However, it is clear that discrete metric is not useful in a practical comparison scenario.  .
.  let us define the set of all unrooted (not necessarily bifurcating) trees with the property that all nodes of degree 1 or 2 are labeled by pairwise disjoint non-empty subsets of
let us define the set of all unrooted (not necessarily bifurcating) trees with the property that all nodes of degree 1 or 2 are labeled by pairwise disjoint non-empty subsets of  . Unlabeled nodes are considered to have
. Unlabeled nodes are considered to have  as their label.
as their label.

 ) and decontraction (denoted
) and decontraction (denoted  ).
).  with the labeling defined by
with the labeling defined by  and an edge
and an edge  we will define the contraction
we will define the contraction  by considering
by considering  and adding a vertex
and adding a vertex  with
with  and edges
and edges  . Thus, we have
. Thus, we have  . The figure below illustrates an example of subsequent contractions on tree in
. The figure below illustrates an example of subsequent contractions on tree in 
 . Let
. Let  where
where  and
and  . Let
. Let  where
where  and
and  . Now, we can construct the tree
. Now, we can construct the tree  and add two vertices
and add two vertices  to it alongside the edge
to it alongside the edge  and redistribute the edges previously incident on
and redistribute the edges previously incident on  . Formally,
. Formally,  with the labeling function defined as
with the labeling function defined as  and
and  for the new nodes. The figure below illustrates two of the possible ways a decontraction can be applied to tree in
for the new nodes. The figure below illustrates two of the possible ways a decontraction can be applied to tree in 
 denote the minimum number of applications of the
denote the minimum number of applications of the  into
into  (whenever we say that two trees are the same we mean that there exists a label preserving isomorphism).
(whenever we say that two trees are the same we mean that there exists a label preserving isomorphism).  within the
within the 
 it follows that we can transform both to the tree with one node, and then apply appropriate inverse transformations, implying that there is a finite (and in fact bounded above by
it follows that we can transform both to the tree with one node, and then apply appropriate inverse transformations, implying that there is a finite (and in fact bounded above by  ) sequence of
) sequence of  operations that transforms
operations that transforms 

 together with a leaf labeling
together with a leaf labeling  that assigns exactly one taxon to each leaf and none to any internal node
that assigns exactly one taxon to each leaf and none to any internal node 
 , i.e. a map
, i.e. a map  (in practice we are interested only in the case
(in practice we are interested only in the case  )
)
 to every leaf is constant.
to every leaf is constant. .
. pairwise distance matrix
pairwise distance matrix  , where we use the notation
, where we use the notation  to denote the path between leaves
to denote the path between leaves  under some norm. One possible choice of the norm is the
under some norm. One possible choice of the norm is the  with the choice
with the choice  proposed by
proposed by  by
by  by
by  and ultrametric space if in addition to the standard properties the metric
and ultrametric space if in addition to the standard properties the metric  . Ultrametric spaces are somewhat bizarre looking since every triangle in such space is isosceles. Furthermore, every point inside an open ball of positive radius is its center, and all open balls of positive radius are
. Ultrametric spaces are somewhat bizarre looking since every triangle in such space is isosceles. Furthermore, every point inside an open ball of positive radius is its center, and all open balls of positive radius are 
 .
.  time, which is a significant speed up when compared to brute-force search over the tree space.
time, which is a significant speed up when compared to brute-force search over the tree space.  we have
we have  . In the case when the distance matrix
. In the case when the distance matrix  , where
, where  is the normalized Hamming distance, as the Jukes-Cantor distance between
is the normalized Hamming distance, as the Jukes-Cantor distance between  where
where  is the estimated Jaccard index.
is the estimated Jaccard index. 


{kind=link}