

As I mentioned in the previous post, I am taking some time off to figure out which direction to pursue next in the blog post series. However, that does not mean that the whole series itself is going into hiatus. In the meantime, I am going to cover a few other topics in phylogenetics. Additionally, I will cross post some of the one-off posts on Dr. Treangen’s lab website. Now, with logistics out of the way let’s get into tree rearrangements.
Introduction
Whenever we are faced with a problem in combinatorial optimization it is often convenient (or even necessary as the majority of interesting problems turn out to be NP-hard) to come up with some heuristic methods. In particular, we often want to explore a “neighborhood” of an object and locate optimal candidates within this neighborhood after which the search can continue from the new local optima. In order to talk about neighborhoods we need to define a metric on the space of objects we are working with (we talked about metric spaces on phylogenetic trees in several previous posts, but in this case we are focusing solely on the tree topology and associated finite metric spaces). In the context of a search problem it is useful to think about the neighborhood of an object as a set of objects that can be obtained from the initial object via some fixed operation. In phylogenetics such operations on trees are typically called tree rearrangements (Felsenstein, 2004). In this post we will take a look at three classical tree rearrangement operations, but keep in mind that there are other ways to mutate trees which lead to different geometries on the tree space.
In the next three sections we will introduce in detail each of the rearrangement operations in the order of increasing neighborhood sizes considered and then briefly discuss the relationships between these three classes, as well as some implications for computational complexity of related problems.
Nearest neighbor interchange
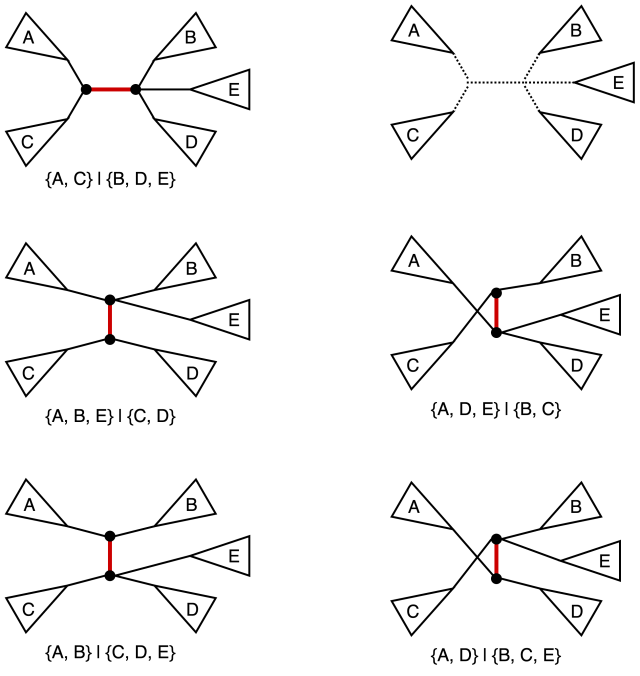
Nearest neighbor interchange (NNI) operation on a tree consists of picking an internal edge (i.e. an edge that is not incident to a leaf) deleting it along with the four edges incident to the vertices of the original edge, after obtaining four components as the result of the deletion there are three distinct ways of reconnecting them back out of which one yields the original tree. It is easy to see that for an unrooted bifurcating tree on n taxa (i.e. a tree with n leaves) there is a total of n-3 internal edges, and hence a total of 2(n-3) NNI-neighbors. We also note that the operation in this classical sense is defined for bifurcating trees (hence the guarantee that the NNI yields for connected components after the edge removal step). The figure below provides an example of NNI and the resulting tree neighbors.

For a small value of n we can reasonably visualize the adjacency structure on the tree space induced by the NNI operation. Namely we can identify non-isomorphic (read: distinct from the point of view of the properties we care about) labeled tree topologies as vertices and let two vertices be connected by an edge iff we can obtain one topology from another via a single NNI.

In general a phylogenetic tree encountered (or I guess inferred) in the wild might not be bifurcating, so it is quite natural to ask how does one generalize the notion of NNI to an arbitrary labeled unrooted tree. At the core of this operation is the process of picking a distinguished edge and then swapping components resulting from an edge deletion operation. Thus, in the case of a multifurcating tree we simply can do the same operation but with a larger set of neighbors being generated. An example is provided in the figure below.
There is some amount of work on the NNI operations and NNI induced metric on phylogenetic trees (Hon and Lam, 1999), although the area tends to feel relatively sparse in terms of the research done.
Subtree prune and regraft
Subtree prune and regraft (SPR) operation on a tree consists of snipping off a branch (which can be an internal or external edge) and then inserting the snipped off subtree onto one of the edges of the remaining tree. This operation is quite well illustrated by its name, especially if you ever had to deal with grafts on actual trees (my grandfather loved to experiment with fruit tree grafting, which to his credit managed to give us a wonderful apricot half-tree on a wild apricot that used to grow in the garden). The figure below is an example of a SPR operation performed on a tree.

It is easy to see that SPR operation results in a larger neighborhood set than the NNI operation. It also useful to note that any tree T’ that can be obtained from a tree T via a single NNI can also be obtained via a single SPR. In other words, if we define the set NNI(T) of trees that can be obtained from T via a single NNI, and the set SPR(T) of trees that can be obtained from T via a single SPR, then the following inclusion holds: NNI(T)⊆SPR(T) (Maddison, 1991). In the spirit of ancient geometers instead of a formalized proof, we will provide the following figure and leave it up to the reader to convince themselves that the inclusion above is indeed true.

Similarly to the NNI case we can compute the exact number of neighbors under the SPR operation for an unrooted bifurcating binary tree on n taxa. The size of this neighborhood similarly to the NNI case is independent of the topology of the tree T and is equal to 2(n-3)(2n-7) with a detailed counting argument provided by Allen and Steel, 2001 on p. 4.
We also note that the way in which we graft a subtree back in will always create an internal node of degree 3, so without a modification to this operation the question of generalizing it to multifurcating trees can be ill-posed.
Tree bisection and reconnection
Tree bisection and reconnection (TBR) operation is the most expansive operation among the three we are considering in this post, in the sense that it yields the largest neighborhoods. TBR consists of splitting the original tree T into two subtrees T’ and T” along a branch and then reconnecting them by joining any two branches of T’ and T” via an edge (in case if one of the subtrees is just a leaf the leaf is simply joined back to the tree via a new branch; in all cases vertices might have to be added to maintain a proper bifurcating tree). The figure below illustrates a TBR operation being performed.

The inclusion we observed for the NNI and SPR neighborhoods extends further and the following holds: NNI(T)⊆SPR(T)⊆TBR(T) (Maddison, 1991). Furthermore, by noting that any TBR can be thought of as a two step process where we first re-attach T” onto the specified edge of T’ and then reposition the attached T’ in order to get correct edge match, we can show that any TBR operation can be replicated by at most two successive SPR operations.
Finally, we note that unlike NNI and SPR, the TBR neighborhood size does depend on the topology of the tree T. However, and upper bound on the size of the neighborhood can be computed and is given by (2n-3)(n-3)2 (Allen and Steel, 2001, p.5).
Induced metrics and computational (in)tractability
We already noted in the introduction that the tree rearrangement operations induce corresponding metrics on the space of phylogenetic trees (more precisely in our current case: the space of unrooted bifurcating tree topologies). We will denote these metrics dNNI(T, T’), dSPR(T, T’), and dTBR(T, T’), respectively. From the inclusion relation on the neighborhoods it immediately follows that for any two unrooted bifurcating trees we have the following inequality: dTBR(T, T’)≤dSPR(T, T’)≤dNNI(T, T’). Furthermore, from our observation in the previous section we also can conclude that dSPR(T, T’)≤2×dTBR(T, T’).
Although we have provided an explicit example of the adjacency graph under NNI metric for the space of unrooted bifurcating trees on 5 taxa, it is not immediately obvious that in the general case the NNI adjacency graph will be connected (i.e. we are not a priori guaranteed to have a connected metric space). However, it turns out that indeed the adjacency graph under the NNI metric is indeed connected, and hence the metric space induced by dNNI(T, T’) is connected. It is easy to see that as a corollary we also get connectedness for the SPR and TBR spaces.
Since the metric spaces are connected and finite we can ask what are their respective diameters (i.e. the smallest distance between two points furthest apart in the space; this is the same definition as the graph diameter). An interesting non-trivial bound on the diameter of the NNI space was derived by Li et al., 1996: authors show that the diameter of the NNI adjacency graph (which we will denote as ΔNNI) is bounded below and above by nlogn terms. More precisely, the following inequality holds: 
However, knowing the diameter of a space does not imply that we can easily compute the distances within it (i.e. shortest paths via NNI, SPR or TBR operations from one tree to another). In fact, the problems of computing NNI, SPR or TBR distance between two arbitrary unrooted bifurcating trees are all NP-hard (NNI: DasGupta et al., 2000; SPR: Bordewich and Semple, 2005, Hickey et al., 2008; TBR: Hein et al., 1996). Allen and Steel, 2001 show that the SPR distance is fixed-parameter tractable, but in general the fixed-parameter tractability results in algorithms that can work efficiently only on small trees or pairs of trees which are relatively close to each other (for a more principled discussion see Whidden and Matsen, 2018). In general, it appears to be a rare case for a metric that is biologically interpretable to be efficiently computable, and vice-a-versa (take for example the Robinson-Foulds distance which is efficiently computable, but not biologically well grounded). There are some recent attempts at developing variants of tree rearrangement operations that have both biological interpretability and are computationally tractable (Collienne and Gavryushkin, 2021).
While we could possibly discuss other tree rearrangement operations or explore the discrete geometries arising from the ones we mentioned in more detail, such work would likely require multiple posts. Thus, we will wrap up here and invite the reader to continue the exploration via following the links provided in the references section.
Data and code availability
All illustrations with the exception of the one from Felsenstein, 2004 were made with draw.io. No additional materials or code were used in this post.
What’s next
I am still deliberating on the exact direction to take for the next stretch of posts, so in the meantime I am intending to continue the one-off posts where I pick a random, but interesting to me, topic in mathematical phylogeny and try to write a coherent text about it. As per usual, if you are interested in me writing about a specific topic do not hesitate to reach out.
References
Allen, Benjamin L., and Mike Steel. “Subtree transfer operations and their induced metrics on evolutionary trees.” Annals of combinatorics 5, no. 1 (2001): 1-15. https://doi.org/10.1007/s00026-001-8006-8
Bordewich, Magnus, and Charles Semple. “On the computational complexity of the rooted subtree prune and regraft distance.” Annals of combinatorics 8, no. 4 (2005): 409-423. https://doi.org/10.1007/s00026-004-0229-z
Collienne, Lena, and Alex Gavryushkin. “Computing nearest neighbour interchange distances between ranked phylogenetic trees.” Journal of Mathematical Biology 82, no. 1 (2021): 1-19. https://doi.org/10.1007/s00285-021-01567-5
Felsenstein, Joseph. Inferring phylogenies. Vol. 2. Sunderland, MA: Sinauer associates, 2004.
Gupta, Bhaskar Das, Xin He, Tao Jiang, Ming Li, and John Tromp. “On computing the nearest neighbor interchange distance.” In Discrete Mathematical Problems with Medical Applications: DIMACS Workshop Discrete Mathematical Problems with Medical Applications, December 8-10, 1999, DIMACS Center, vol. 55, p. 125. American Mathematical Soc., 2000. PDF on author’s page
Hein, Jotun, Tao Jiang, Lusheng Wang, and Kaizhong Zhang. “On the complexity of comparing evolutionary trees.” Discrete Applied Mathematics 71, no. 1-3 (1996): 153-169. https://doi.org/10.1016/S0166-218X(96)00062-5
Hickey, Glenn, Frank Dehne, Andrew Rau-Chaplin, and Christian Blouin. “SPR distance computation for unrooted trees.” Evolutionary Bioinformatics 4 (2008): EBO-S419. https://doi.org/10.4137/EBO.S419
Hon, Wing-Kai, and Tak-Wah Lam. “Approximating the nearest neighbor interchange distance for evolutionary trees with non-uniform degrees.” In International Computing and Combinatorics Conference, pp. 61-70. Springer, Berlin, Heidelberg, 1999. https://doi.org/10.1007/3-540-48686-0_6
Li, Ming, John Tromp, and Louxin Zhang. “On the nearest neighbour interchange distance between evolutionary trees.” Journal of Theoretical Biology 182, no. 4 (1996): 463-467. https://doi.org/10.1006/jtbi.1996.0188
Maddison, David R. “The discovery and importance of multiple islands of most-parsimonious trees.” Systematic Biology 40, no. 3 (1991): 315-328. https://doi.org/10.1093/sysbio/40.3.315
 between points
between points  and
and  . Choose a set of points
. Choose a set of points  such that
such that  . Note that it follows from the definition of a local geodesic that
. Note that it follows from the definition of a local geodesic that  is a geodesic. Now, assume by induction that
is a geodesic. Now, assume by induction that  is a geodesic for all $j\leq i$. Hence, we have
is a geodesic for all $j\leq i$. Hence, we have  by the induction hypothesis and
by the induction hypothesis and  as noted above. Consider a corresponding model triangle
as noted above. Consider a corresponding model triangle  in Euclidean space (if you forgot what a model triangle is check:
in Euclidean space (if you forgot what a model triangle is check:  be a point on
be a point on  such that
such that  . Since we are working in a CAT(0) space it follows that
. Since we are working in a CAT(0) space it follows that  . On the other hand, by induction hypothesis we have
. On the other hand, by induction hypothesis we have  . Combining this result with triangle inequality gives us
. Combining this result with triangle inequality gives us  , and hence
, and hence  and therefore
and therefore  . From here we can conclude that
. From here we can conclude that  is a geodesic and hence $\Gamma_\varepsilon$ is a geodesic.
is a geodesic and hence $\Gamma_\varepsilon$ is a geodesic. between which we are computing geodesics have distinct edges. We will also call two sets of edges
between which we are computing geodesics have distinct edges. We will also call two sets of edges  and
and  compatible if all pairs of splits associated with these edges are compatible, or equivalently if the union
compatible if all pairs of splits associated with these edges are compatible, or equivalently if the union  defines a tree. In other words, compatible sets of edges produce “intermediate” trees between
defines a tree. In other words, compatible sets of edges produce “intermediate” trees between  and
and  . Now, for a pair of partitions
. Now, for a pair of partitions  ,
,  of
of  and
and  , respectively, we can define an associated path space given that the following property holds: for every
, respectively, we can define an associated path space given that the following property holds: for every  ,
,  and
and  are compatible. To construct the path space consider the collection of orthants
are compatible. To construct the path space consider the collection of orthants  given by
given by  , where
, where  is the mapping of a tree to the corresponding orthant. We will call the corresponding partition pair
is the mapping of a tree to the corresponding orthant. We will call the corresponding partition pair  the support, and the shortest path between
the support, and the shortest path between  the path space geodesic. These definitions make the geodesic in the tree space a more concrete and constructable object, which of course is important for any algorithmic approach. Furthermore, the following theorem from
the path space geodesic. These definitions make the geodesic in the tree space a more concrete and constructable object, which of course is important for any algorithmic approach. Furthermore, the following theorem from  .
. be a geodesic between
be a geodesic between  latex \mathcal{B}=\{B_1,…,B_k\} of
latex \mathcal{B}=\{B_1,…,B_k\} of  .
. -path
-path  in the support there is no nontrivial partition
in the support there is no nontrivial partition  ,
,  such that
such that  is compatible with
is compatible with  and
and  .
. and the corresponding path is given by uniform contraction of all edges which results in star tree and then a follow up uniform decontraction. Now, the algorithm will proceed iteratively at each step checking whether the condition in Theorem 3 is satisfied and if not proposing a new support and a new proper path.
and the corresponding path is given by uniform contraction of all edges which results in star tree and then a follow up uniform decontraction. Now, the algorithm will proceed iteratively at each step checking whether the condition in Theorem 3 is satisfied and if not proposing a new support and a new proper path.  latex A\subseteq E(T), B\subseteq E(T^\prime)$ as a bipartite graph with vertices given by
latex A\subseteq E(T), B\subseteq E(T^\prime)$ as a bipartite graph with vertices given by  where the splits associated with
where the splits associated with  and
and  are incompatible. It is easy to see that for any
are incompatible. It is easy to see that for any  compatibility of
compatibility of  and
and  is equivalent to
is equivalent to  ,
,  such that the vertex set
such that the vertex set  is independent in
is independent in  and
and  satisfies the above condition.
satisfies the above condition. . Further analysis of the algorithm steps yields that the total runtime to find the geodesic is
. Further analysis of the algorithm steps yields that the total runtime to find the geodesic is  . For the exact details of the proof of correctness of the algorithm and the runtime I suggest reading the original manuscript by
. For the exact details of the proof of correctness of the algorithm and the runtime I suggest reading the original manuscript by  into some metric space
into some metric space  . Thus, by associating every tree in the tree space with some point in the chosen metric space, we induce a pseudometric on tree space. However, such embeddings/parameterizations are not guaranteed to be “nice” with respect to the questions we aim to answer. Namely, not all embeddings are injective (multiple distinct trees can end up being mapped to the same point in
. Thus, by associating every tree in the tree space with some point in the chosen metric space, we induce a pseudometric on tree space. However, such embeddings/parameterizations are not guaranteed to be “nice” with respect to the questions we aim to answer. Namely, not all embeddings are injective (multiple distinct trees can end up being mapped to the same point in  , hence in general we induce a pseudometric rather than a metric via embedding) although in the case of
, hence in general we induce a pseudometric rather than a metric via embedding) although in the case of  that would enable fruitful analyses. From now onwards (unless otherwise specified) we will assume that the embedding
that would enable fruitful analyses. From now onwards (unless otherwise specified) we will assume that the embedding  is injective.
is injective.  is path-connected in
is path-connected in  , where $\tau_i$ is the time difference between the i-th and i+1-th nodes. The picture below taken from
, where $\tau_i$ is the time difference between the i-th and i+1-th nodes. The picture below taken from 
 that embeds the space of ultrametric phylogenetic trees into a disjoint union of
that embeds the space of ultrametric phylogenetic trees into a disjoint union of  (
( ). We will impose an upper bound on all orthants to turn our construction into a cubical complex for the ease of the exposition. However, the construction of 𝛕-space with orthants will still yield a CAT(0) space with the key properties and desiderata (D1)-(D5′) preserved.
). We will impose an upper bound on all orthants to turn our construction into a cubical complex for the ease of the exposition. However, the construction of 𝛕-space with orthants will still yield a CAT(0) space with the key properties and desiderata (D1)-(D5′) preserved.
 face. Since the resulting space is a cubical complex, we can naturally consider the Euclidean metric within each cube with paths for joining points in different cubes being the sums of the respective within cube paths.
face. Since the resulting space is a cubical complex, we can naturally consider the Euclidean metric within each cube with paths for joining points in different cubes being the sums of the respective within cube paths. , then the resulting facet is only contained in the cube of corresponding rooted topology. If setting
, then the resulting facet is only contained in the cube of corresponding rooted topology. If setting  does not result in a multifurcating topology then the corresponding facet will be shared by exactly two cubes (see example in Figure 2 and/or Figure 2 of
does not result in a multifurcating topology then the corresponding facet will be shared by exactly two cubes (see example in Figure 2 and/or Figure 2 of  with the intrinsic Euclidean metric is CAT(0) if and only if
with the intrinsic Euclidean metric is CAT(0) if and only if  : if three
: if three  -cubes of
-cubes of  -cubes, then they are contained within a
-cubes, then they are contained within a  -cube in
-cube in  . Hence, there exists
. Hence, there exists  s.t. it is greater than zero in both of the first two cubes, and is zero in their shared (k+1)-cube. Now, if the first and third cube share a (k+1)-cube then it follows that their i and r coordinates both have to be zero in the shared cube, implying that the s coordinate has to be resolved in the same way between the first and third cube. However, the same exact argument can be repeated for the second and third cube implying that the first and second cubes had to be indetical.
s.t. it is greater than zero in both of the first two cubes, and is zero in their shared (k+1)-cube. Now, if the first and third cube share a (k+1)-cube then it follows that their i and r coordinates both have to be zero in the shared cube, implying that the s coordinate has to be resolved in the same way between the first and third cube. However, the same exact argument can be repeated for the second and third cube implying that the first and second cubes had to be indetical.
 a geodesic from
a geodesic from  to
to  is the map
is the map ![c:[0, l]\to X](https://s0.wp.com/latex.php?latex=c%3A%5B0%2C+l%5D%5Cto+X&bg=fafafa&fg=6c6c8c&s=0&c=20201002) s.t.
s.t.  and
and  . We will call a metric space a geodesic space if for any
. We will call a metric space a geodesic space if for any  there exists a geodesic from
there exists a geodesic from  we have the geodesic given by
we have the geodesic given by  . On a sphere geodesic between two points
. On a sphere geodesic between two points  is the arc segment obtained by intersecting a plane through
is the arc segment obtained by intersecting a plane through 
 is a set of tree points
is a set of tree points  called vertices and a choice of three geodesic segments
called vertices and a choice of three geodesic segments ![[p, q], [q, r], [r, p]](https://s0.wp.com/latex.php?latex=%5Bp%2C+q%5D%2C+%5Bq%2C+r%5D%2C+%5Br%2C+p%5D&bg=fafafa&fg=6c6c8c&s=0&c=20201002) joining them called sides, we will denote such a triangle
joining them called sides, we will denote such a triangle ![\Delta([p, q], [q, r], [r, p])](https://s0.wp.com/latex.php?latex=%5CDelta%28%5Bp%2C+q%5D%2C+%5Bq%2C+r%5D%2C+%5Br%2C+p%5D%29&bg=fafafa&fg=6c6c8c&s=0&c=20201002) or more briefly
or more briefly  . Note, that the later notation is not precise, as in the case of a non-uniquely geodesic space there is not necessarily a unique choice of a geodesic between two vertices. Finally, we will write
. Note, that the later notation is not precise, as in the case of a non-uniquely geodesic space there is not necessarily a unique choice of a geodesic between two vertices. Finally, we will write  to indicate that
to indicate that ![[p, q]\cup [q, r]\cup [r, p]](https://s0.wp.com/latex.php?latex=%5Bp%2C+q%5D%5Ccup+%5Bq%2C+r%5D%5Ccup+%5Br%2C+p%5D&bg=fafafa&fg=6c6c8c&s=0&c=20201002) .
.  Euclidean n-dimensional space,
Euclidean n-dimensional space,  the n-sphere, and
the n-sphere, and  hyperbolic n-space. We already defined geodesics in the case of
hyperbolic n-space. We already defined geodesics in the case of  . For the n-sphere we will define the metric via cosine distance
. For the n-sphere we will define the metric via cosine distance  where the inner product is in the corresponding embedding
where the inner product is in the corresponding embedding  and the corresponding geodesics are given by minimal great arcs (for full definition and description see
and the corresponding geodesics are given by minimal great arcs (for full definition and description see  which consists of
which consists of  with bilinear form
with bilinear form  and defining
and defining  . The distance on this space will be given by
. The distance on this space will be given by  (similarly to the sphere case, see
(similarly to the sphere case, see  is a triangle in the model space
is a triangle in the model space  that satisfies
that satisfies  ,
,  , and
, and  . A point
. A point ![\overline{x}\in[\overline{p}, \overline{q}]](https://s0.wp.com/latex.php?latex=%5Coverline%7Bx%7D%5Cin%5B%5Coverline%7Bp%7D%2C+%5Coverline%7Bq%7D%5D&bg=fafafa&fg=6c6c8c&s=0&c=20201002) for
for ![x\in[p, q]](https://s0.wp.com/latex.php?latex=x%5Cin%5Bp%2C+q%5D&bg=fafafa&fg=6c6c8c&s=0&c=20201002) is called a comparison point if
is called a comparison point if  . Note, that for
. Note, that for  we need an additional condition on the perimeter of a triangle to guarantee existence of a comparison triangle, for the purposes of this post we will state the condition, but not elaborate on it in detail.
we need an additional condition on the perimeter of a triangle to guarantee existence of a comparison triangle, for the purposes of this post we will state the condition, but not elaborate on it in detail.  be a metric space and let
be a metric space and let  (the diameter of the
(the diameter of the  satisfies the CAT(k) inequality if for the corresponding comparison triangle
satisfies the CAT(k) inequality if for the corresponding comparison triangle  and all
and all  with corresponding comparison points
with corresponding comparison points  the inequality
the inequality  is satisfied. Thus, for a
is satisfied. Thus, for a  we will call
we will call  -geodesic and only requiring the inequality condition for triangles of perimeter bounded by
-geodesic and only requiring the inequality condition for triangles of perimeter bounded by 
![I=[0, 1]](https://s0.wp.com/latex.php?latex=I%3D%5B0%2C+1%5D&bg=fafafa&fg=6c6c8c&s=0&c=20201002) be the unit interval, then we call the n-fold product
be the unit interval, then we call the n-fold product  the unit cube. We will let
the unit cube. We will let  denote a point by convention. Since we will be mainly operating in
denote a point by convention. Since we will be mainly operating in  dimensions, we will use the term “face” to describe any-dimensional face of the cube. Thus, the faces of
dimensions, we will use the term “face” to describe any-dimensional face of the cube. Thus, the faces of ![[0, 1]](https://s0.wp.com/latex.php?latex=%5B0%2C+1%5D&bg=fafafa&fg=6c6c8c&s=0&c=20201002) (the 1-dimensional face). Analogously, for
(the 1-dimensional face). Analogously, for  which can be written as
which can be written as  where each
where each  is a face of
is a face of  . The dimension of the face will be the sum of dimensions of
. The dimension of the face will be the sum of dimensions of  .
.  by an equivalence relation
by an equivalence relation  with the restrictions
with the restrictions  of the natural projection
of the natural projection  satisfying
satisfying the map
the map  is injective;
is injective; then there is an isometry
then there is an isometry  from a face
from a face  onto a face
onto a face  such that
such that  .
.
 error).
error). is a function
is a function  satisfying
satisfying


 . Recall from the previous post that in the case when the third condition (called triangle inequality) is replaced by a stricter condition
. Recall from the previous post that in the case when the third condition (called triangle inequality) is replaced by a stricter condition  we obtain an ultrametric space.
we obtain an ultrametric space.  if
if  and
and  otherwise. This metric is called the discrete metric (and it is trivial to check that discrete metric is also an ultrametric). Discrete metric provides us with an important realization that any set can be turned into a metric space. However, it is clear that discrete metric is not useful in a practical comparison scenario.
otherwise. This metric is called the discrete metric (and it is trivial to check that discrete metric is also an ultrametric). Discrete metric provides us with an important realization that any set can be turned into a metric space. However, it is clear that discrete metric is not useful in a practical comparison scenario.  .
.  let us define the set of all unrooted (not necessarily bifurcating) trees with the property that all nodes of degree 1 or 2 are labeled by pairwise disjoint non-empty subsets of
let us define the set of all unrooted (not necessarily bifurcating) trees with the property that all nodes of degree 1 or 2 are labeled by pairwise disjoint non-empty subsets of  . Unlabeled nodes are considered to have
. Unlabeled nodes are considered to have  as their label.
as their label.

 ) and decontraction (denoted
) and decontraction (denoted  ).
).  with the labeling defined by
with the labeling defined by  and an edge
and an edge  we will define the contraction
we will define the contraction  by considering
by considering  and adding a vertex
and adding a vertex  with
with  and edges
and edges  . Thus, we have
. Thus, we have  . The figure below illustrates an example of subsequent contractions on tree in
. The figure below illustrates an example of subsequent contractions on tree in 
 . Let
. Let  where
where  and
and  . Let
. Let  where
where  and
and  . Now, we can construct the tree
. Now, we can construct the tree  and add two vertices
and add two vertices  to it alongside the edge
to it alongside the edge  and redistribute the edges previously incident on
and redistribute the edges previously incident on  . Formally,
. Formally,  with the labeling function defined as
with the labeling function defined as  and
and  for the new nodes. The figure below illustrates two of the possible ways a decontraction can be applied to tree in
for the new nodes. The figure below illustrates two of the possible ways a decontraction can be applied to tree in 
 denote the minimum number of applications of the
denote the minimum number of applications of the  into
into  (whenever we say that two trees are the same we mean that there exists a label preserving isomorphism).
(whenever we say that two trees are the same we mean that there exists a label preserving isomorphism).  within the
within the 
 it follows that we can transform both to the tree with one node, and then apply appropriate inverse transformations, implying that there is a finite (and in fact bounded above by
it follows that we can transform both to the tree with one node, and then apply appropriate inverse transformations, implying that there is a finite (and in fact bounded above by  ) sequence of
) sequence of  operations that transforms
operations that transforms 

 with no cycles and in which
with no cycles and in which  together with a leaf labeling
together with a leaf labeling  that assigns exactly one taxon to each leaf and none to any internal node
that assigns exactly one taxon to each leaf and none to any internal node 
 , i.e. a map
, i.e. a map  (in practice we are interested only in the case
(in practice we are interested only in the case  )
)
 to every leaf is constant.
to every leaf is constant. taxa is
taxa is  .
. pairwise distance matrix
pairwise distance matrix  , where we use the notation
, where we use the notation  to denote the path between leaves
to denote the path between leaves  under some norm. One possible choice of the norm is the
under some norm. One possible choice of the norm is the  with the choice
with the choice  proposed by
proposed by  by
by  by
by  and ultrametric space if in addition to the standard properties the metric
and ultrametric space if in addition to the standard properties the metric  . Ultrametric spaces are somewhat bizarre looking since every triangle in such space is isosceles. Furthermore, every point inside an open ball of positive radius is its center, and all open balls of positive radius are
. Ultrametric spaces are somewhat bizarre looking since every triangle in such space is isosceles. Furthermore, every point inside an open ball of positive radius is its center, and all open balls of positive radius are 
 .
.  time, which is a significant speed up when compared to brute-force search over the tree space.
time, which is a significant speed up when compared to brute-force search over the tree space.  we have
we have  . In the case when the distance matrix
. In the case when the distance matrix  , where
, where  is the normalized Hamming distance, as the Jukes-Cantor distance between
is the normalized Hamming distance, as the Jukes-Cantor distance between  where
where  is the estimated Jaccard index.
is the estimated Jaccard index. 


{kind=link}